
Assaying cell cycle status using flow cytometry
Kang Ho Kim
1,*
and Joel M. Sederstrom
2
1
Department of Molecular and Cellular Biology, Baylor College of Medicine, 1 Baylor Plaza,
Houston TX 77030, USA
2
Cytometry and Cell Sorting Core, Baylor College of Medicine, 1 Baylor Plaza, Houston TX
77030, USA
Abstract
In this unit, we describe two protocols for analyzing cell cycle status using flow cytometry. The
first is based on the simultaneous analysis of proliferation specific marker (Ki-67) and cellular
DNA content, which discriminates resting/quiescent cell populations (G0 cell) and quantifies cell
cycle distribution (G1, S or G2/M, respectively). The second is based on differential staining of
DNA and RNA through co-staining of Hoechst 33342 and Pyronin Y, which is also useful to
identify G0 cells from G1 cells. Along with these methods for analyzing cell cycle status, we
outline the basics of two additional methods for cell proliferation assays and recent updates of
newly-developed fluorophores, which allows multiplex analysis of cell cycle status, cell
proliferation and a gene of interest using flow cytometry.
Keywords
Cell Cycle; Flow Cytometry; Ki-67; Propidium Iodide; Pyronin Y; Hoechst 33342
UNIT INTRODUCTION
Assessing cell cycle distribution and cell proliferation is important for studying cell growth
differentiation, senescence and apoptosis. This enables one to investigate underlying basic
mechanisms as well as to evaluate therapeutic efficacies of anti-cancer drugs. During cell
cycle progression, proliferating cells sequentially undergo a transition of G1→S→G2→M
phases for synthesis of DNA, preparation of cell division and subsequent mitosis process
(Malumbres and Barbacid, 2009). However, under certain circumstances, cells can enter G0
phase, where the cells are neither dividing nor preparing for proliferation. These resting cells
are characterized by having minimal cell cycle machinery and maintaining specialized
*
Correspondence to; Kang Ho Kim, Ph.D., Department of Molecular and Cellular Biology, Baylor College of Medicine, 1 Baylor
Plaza, Houston TX, 77030, USA, Tel: (713) 798-6223, [email protected].
Conflict of Interest
The Authors declare no conflict of interest.
INTERNET RESOURCES
http://www.lifetechnologies.com/us/en/home/life-science/cell-analysis/flow-cytometry/cell-health-and-viability-assays-for-flow-
cytometry/cell-proliferation-assays-for-flow-cytometry/click-it-edu-cell-proliferation-assay-kits-for-flow-cytometry.html
Web site for the introduction of Click-iT® Plus Assay Kits
HHS Public Access
Author manuscript
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Published in final edited form as:
Curr Protoc Mol Biol. ; 111: 28.6.1–28.6.11. doi:10.1002/0471142727.mb2806s111.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

cellular functions rather than by proceeding to cell proliferation. This resting state is also
referred as quiescent (Zetterberg et al., 1995).
The earliest and simplest approach to analyze cell cycle status is to measure cellular DNA
content at a single time point (Darzynkiewicz and Huang, 2004). This reveals a snapshot of
cell cycle status among 3 distinct groups (i.e., G0/G1 (2n), S (2n~4n), and G2/M (4n) phase,
respectively). In 1969, the feulgen-DNA staining method was first described to analyze cell
cycle distribution (Van Dilla et al., 1969). Since then, many fluorescent DNA dyes have
been developed for multiplex analysis of cellular DNA content and other proliferation-
related markers (Table 1). However, this method is insufficient to understand detailed cell
cycle status because DNA content alone cannot distinguish resting/quiescent cells (G0) from
G1 phase cells.
To overcome this limitation, some alternative methods have been developed. First, resting/
quiescent and proliferating cell fractions can be identified by proliferation-associated
proteins and/or nuclear proliferation antigens such as Ki-67 and proliferating cell nuclear
antigen (PCNA). Ki-67 antigen is rarely detected in G0 phase, highly expressed in the
nuclear region of proliferating cells (maximum in G2 and early M phases) and rapidly
degraded during anaphase and telophase of mitosis processes (Gerdes et al., 1984).
Likewise, PCNA is a good marker for proliferating cells and is concentrated in S phase
(Kurki et al., 1986), which is useful to separate S phase cells. Second, quantification of
intracellular RNA by Hoechst 33342/Pyronin Y double staining can be an alternative way to
study cell cycle status because highly proliferating cells usually contain higher levels of
RNA compared to resting/quiescent cells. Historically, Pyronin Y has been widely used for
microscopic observation of cellular RNA in combination with methyl green (Scott, 1967).
Its application was extended to flow cytometry by Howard Shapiro in 1981 and further
defined by Zibgniew Darzynkiewicz in 2004 (Darzynkiewicz et al., 2004; Shapiro, 1981). In
this unit, two basic flow cytometric techniques are described for assessing cell cycle status
through costaining of Ki-67/DNA (Basic Protocol 1) and quantification of intracellular RNA
(Basic Protocol 2).
BASIC PROTOCOL 1
Title
Flow cytometric analysis of Ki-67 and DNA content for analyzing cell cycle status.
Introduction
The Ki-67 antibody was first described to recognize a nuclear protein only present in
proliferating cells (Gerdes et al., 1983). Later, the nuclear antigen detected by Ki-67
antibody was identified as two isoforms of 320 kDa and 359 kDa Ki-67 protein, which may
be necessary for maintenance of cell proliferation (Schluter et al., 1993). The Ki-67-positive
population is mainly limited to proliferating cells in many cell types during active phases of
the cell cycle (G1, S, G2 and M phases), whereas it is absent from resting/quiescent cells
(Gerdes et al., 1984; Schwarting et al., 1986). Thus, Ki-67 has been extensively used to
predict the growth rate of many cancer samples from human patients. This protocol provides
Kim and Sederstrom
Page 2
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

a detailed procedure for determining cell cycle status of tissue culture cells through double
staining of Ki-67 and PI using flow cytometry.
Materials
Solutions and reagents—1X Phosphate buffered saline (PBS)
70% Cold ethanol (−20°C)
FACS buffer (see recipe)
PI staining solution (see recipe)
FITC-conjugated Ki-67 antibody
NOTE: Alternatively, various fluorescent dyes such as PE and APC can be used for Ki-67 in
combination with other DNA-binding fluorescent dyes (Table 1) in order to avoid
significant spectral overlap.
Special equipment—Flow cytometer equipped with 488 nm blue laser and appropriate
filter sets detecting FITC and PI fluorescence.
Steps and Annotations
Harvest, fix and permeabilize cells
1. Plate cells at proper density so that cells should not be confluent at the time of cell
harvest (See Critical Parameters).
2. Harvest and pellet cells (1 × 10
6
) after washing with 10 ml PBS by centrifuging 5
min at 200 × g.
3. Remove supernatant and resuspend cells in 0.5 ml PBS
4. Add 4.5 ml pre-chilled 70% cold ethanol (−20°C) in a drop wise manner to the cell
suspension while vortexing.
In this step, cell clumping should be minimized (see Troubleshooting).
5. Incubate the fixed cells at least 2 hour at −20°C. Cells may be stored in ethanol
fixative for several weeks at −20°C prior to antibody staining.
Stain cells with Ki-67 antibody and fluorescent DNA dyes
6. Centrifuge 3 min at 300 × g and remove ethanol.
7. Rinse cells with 5 ml FACS buffer tw1ice by centrifuging 5 min at 200 × g.
8.Remove supernatant and resuspend cells in 100 μl FACS buffer (1 × 10
6
cells/100 μl).
9. Add 10 μl pre-diluted Ki-67-FITC antibody and incubate 30 min at room
temperature.
Kim and Sederstrom
Page 3
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Refer to manufacturer's instruction for optimal antibody dilution. For the best quality of
positive cell discrimination from negative cells, titration of Ki-67-FITC antibody is
required if nothing is specified.
After this step, the rest of the procedure should be performed in the dark.
10. Wash with 5 ml FACS buffer twice by centrifuging 5min at 200 × g.
11. Remove supernatant. Add 500 μl PI staining solution and resuspend pellet gently.
12. Incubate 20 min at room temperature.
Washing is not necessary.
Perform flow cytometry
13. Set up and adjust flow cytometer with a blue laser (488 nm) and detection filters
(530/30 nm band pass for FITC and 610/20 nm band pass for PI).
Ki-67-FITC signal in logarithmic mode and PI signal in linear mode. PI fluorescence
can be detected in 585/42 nm band pass, 670 nm long pass filters or something capable
of detecting PI fluorescence.
14. Set a low flow rate (less than 400 events/second) for optimal resolution of PI
fluorescence.
15. Exclude doublets by creating a combination of same-channel bivariate plots
utilizing Area vs Height or Area vs Width (i.e., FSC, SSC and PI fluorescence).
Singlet events are presented in a diagonal pattern. Doublets have lower Height and
higher Width values.
16. Acquire the fluorescence and analyze cell cycle stages of each sample (See
Anticipated Results).
Appropriate Compensation procedures between fluorophores should be utilized.
ALTERNATE PROTOCOL 1
Title
Simultaneous staining of cell surface antigens with Ki-67 and PI.
Introduction
This method can be used for multiplex staining of surface proteins and Ki-67/DNA, which
enables analysis of surrogate markers for resting/quiescent cells. After first staining of
surface antigen, both fixation and permeabilization are subsequently required. Here, we
describe classical procedures of surface antigen staining followed by fixation (4%
formaldehyde) and permeabilization (1% saponin). The saponin-based permeabilization is a
reversible process so the remainder of the procedure should be performed in a presence of
saponin. This method usually preserves cell shape as well as structure of cellular
components but takes longer time for surface antigen staining, fixation and permeabilization
processes.
Kim and Sederstrom
Page 4
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Material List
Solutions and reagents—1X Phosphate buffered saline (PBS)
FACS buffer (see recipe)
Fluorophore-conjugated antibody against surface antigen
Fixation solution (4% paraformaldehyde)
Permeabilization solution (see recipe)
Saponin wash buffer (see recipe)
FITC-conjugated Ki-67 antibody
PI/saponin staining solution (see recipe)
NOTE: Other fixatives and permeabilization buffers are commercially available (FIX &
PERM® Cell Fixation & Cell Permeabiliazation Kit (#GAS003/GAS004) from Life
Technologies; Cytofix/Cytoperm™ Fixation/Permeabiliazation Solution Kit (#554714) from
BD Biosciences, etc). For detailed procedures, refer to manufacturer's instructions.
NOTE: Optimization of paraformaldehyde (1~4%) and saponin (0.1~1%) concentrations are
needed.
Special equipment—Flow cytometer equipped with 488 nm blue laser and appropriate
filter sets for detecting FITC and PI fluorescence. Depending on the fluorophore for surface
antigens, additional laser and filter sets are needed.
Steps and Annotations
Stain cell surface antigen with fluorescent-conjugated antibody
1. Harvest cells (1 × 10
6
) and wash with 10 ml PBS by centrifuging 5 min at 200 × g.
2. Remove supernatant and resuspend cells in 100 μl FACS buffer.
3. Add non-fixation sensitive fluorophore-conjugated primary antibody detecting cell
surface antigen and incubate 20 min in the dark at 4°C.
The detection wavelengths for the fluorescent dyes for surface antigens should not
overlap with those for FITC and PI. For optimal antibody dilution, refer to the
manufacturer's instructions or perform a titration. After this step, the rest of the
procedure should be done in the dark.
4. Wash with 5 ml FACS buffer twice by centrifuging 5 min at 200 × g.
Fix and permeabilize cells for intracellular staining
5. Resuspend cells in 200 μl Fixation solution (4% paraformaldehyde).
6. Incubate 20 min at room temperature.
Kim and Sederstrom
Page 5
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

7. Add 5 ml PBS and centrifuge 5 min at 200 × g to remove fixative.
8. Resuspend cells in 200 μl Permeabilization solution and incubate 20 min at room
temperature.
After this step, 0.5% saponin should be present in all buffers used in this protocol.
9. Wash cells with 5 ml Saponin wash buffer and centrifuge 5 min at 200 × g.
Stain with Ki-67 and PI
10. Resuspend cells in 100 μl Saponin wash buffer and add 10 μl pre-diluted Ki-67-
FITC antibody.
Refer to manufacturer's instruction for optimal antibody dilution. For the best quality of
positive cell discrimination from negative cells, titration of Ki-67-FITC antibody is
required
11. Incubate 30 min at room temperature.
12. Wash cells with 5 ml Saponin wash buffer twice by centrifuging 5 min at 200 × g.
13. Add 500 μl PI/saponin staining solution and resuspend pellet gently.
14. Incubate 20 min at room temperature.
Washing is not necessary.
Perform flow cytometry
15. Set up and adjust flow cytometer with proper laser and filter sets
For detecting FITC and PI, a blue laser (488 nm), detection filters of 530/30 nm band
pass (for FITC) and 610/20 nm band pass (for PI) are used. Alternatively, 585/42 nm
band pass, 670 nm long pass filters or something capable of detecting PI fluorescence
can be used. For detecting surface staining, use proper laser and filter sets according to
the selected fluorophore. Ki-67-FITC signal in logarithmic mode and PI signal in linear
mode.
16. Set a low flow rate (less than 400 events/second) for optimal resolution of PI
fluorescence.
17. Exclude doublets by creating a combination of same-channel bivariate plots
utilizing Area vs Height or Area vs Width (i.e., FSC, SSC and PI fluorescence).
Singlet events are presented in a diagonal pattern. Doublets have lower Height and
higher Width values.
18. Acquire the fluorescence and analyze cell cycle stages of each sample.
Appropriate Compensation procedures between fluorophores should be utilized.
BASIC PROTOCOL 2
Title
Pyronin Y and Hoechst 33342 staining for analyzing cell cycle status.
Kim and Sederstrom
Page 6
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Introduction
The other way to identify the resting cells (G0 cells) from proliferating cell is to determine
the total RNA content inside the cells. Generally, resting/quiescent cells at G0 phase have
lower levels of RNA compared with proliferating interphase cells (G1-S-G2-M phase). To
address this, double staining of Hoechst 33342 and Pyronin Y is widely used. Pyronin Y
intercalates both double stranded DNA and double stranded RNA, which can be used for
visualization of RNA as an orange-red band during electrophoresis. In the presence of DNA-
chelating fluorescent dye such as Hoechst 33342, interactions of Pyronin Y and DNA
complex are disrupted and Pyronin Y mainly stains RNA (Shapiro, 1981), allowing the
quantification of RNA amount in a single cell level. Here, we describe a basic protocol for
double staining of cells with Pyronin Y and Hoechst 33342 to dissect resting and
proliferating cells.
Material List
Solutions and reagents—1× Phosphate buffered saline (PBS)
70% Cold ethanol (−20°C)
FACS buffer (see recipe)
Hoechst/PY staining solution (see recipe)
Special equipment—Flow cytometer equipped with both 355 nm UV and 488 nm blue
laser to activate Hoechst 33342 and Pyronin Y. 488 nm laser can be replaced by 532 nm
green or 561 nm yellow-green lasers. Appropriate filter sets are needed.
Steps and Annotations
1. Harvest cells (1 × 10
6
) and wash with 10 ml PBS by centrifuging 5 min at 200 × g.
2. Resuspend cells in 0.5 ml PBS.
3. Fix cells by adding 4.5 ml pre-chilled 70% ethanol (−20°C) drop wise while
vortexing
4. Incubate at least 2 hour at −20°C.
In this step, cell clumping should be minimized (see Troubleshooting). Ethanol-fixed
cells can be stored several weeks at −20°C.
5. Eliminate residual ethanol by centrifuging 3 min at 300 × g. Remove and discard
supernatant.
6. Wash cells with 5 ml FACS buffer twice by centrifuging 5 min at 200 × g.
7. Stain cells using 0.5 ml Hoechst/PY staining solution.
The sample should be kept in the dark. A washing step is not necessary.
8. Incubate 20 min at room temperature and analyze the fluorescences in flow
cytometry.
Kim and Sederstrom
Page 7
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Perform flow cytometry
9. Set up and adjust flow cytometer with UV (355 nm) and blue (488 nm) lasers as well
as proper filter sets (450/50 nm band pass for Hoechst and 575/26 nm band pass for
Pyronin Y)
Pyronin Y is excited by 488 nm blue laser and 532 or 561 nm lasers are also available.
Maximal emission wavelength of Hoechst 33342 is 461 nm and that of Pyronin Y is
575 nm. Both Hoechst 33342 and Pyronin Y signal in a linear mode.
10. Exclude doublets by creating a combination of same-channel bivariate plots
utilizing Area vs Height or Area vs Width (i.e., FSC, SSC and Hoechst fluorescence).
Singlet events are presented in a diagonal pattern. Doublets have lower Height and
higher Width values.
11. Acquire the fluorescence and analyze cell cycle stages of each sample (See
Anticipated Results).
Appropriate Compensation procedures between fluorophores should be utilized.
REAGENTS AND SOLUTIONS
FACS buffer
1X PBS supplemented with:
2% (v/v) heat-inactivated, sterile-filtered fetal bovine serum (10 ml FBS per 500 ml)
1 mM EDTA (1 ml of 0.5 M EDTA stock per 500 ml)
Store at 4°C for up to 6 months. 2% (v/v) FBS can be replaced by 0.2~0.5% (w/v) bovine
serum albumin (BSA). Sodium azide (NaN
3
, 0.1%) can be added to prevent microbial
contamination.
PI staining solution
1X PBS supplemented with:
50 μg/ml propidium iodide (50 μl of 1 mg/ml PI stock per 1 ml)
100 μg/ml RNase (10 μl of 10 mg/ml RNase stock per 1ml)
2 mM MgCl
2
(2 μl of 1 M MgCl
2
stock per 1 ml)
Prepare freshly and keep in the dark at 4°C before use.
Permeabilization solution
10 mM HEPES buffer, pH 7.2 (10 μl of 1 M HEPES buffer stock per 1 ml)
1% (w/v) saponin (100 μl of 10% (w/v) saponin stock per 1 ml)
Prepare freshly and store at 4°C before use.
Kim and Sederstrom
Page 8
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Saponin wash buffer
10 mM HEPES buffer, pH 7.2 (10 μl of 1 M HEPES buffer stock per 1 ml)
0.5% (w/v) saponin (50 μl of 10% (w/v) saponin stock per 1 ml)
Prepare freshly and store at 4°C before use.
PI/saponin staining solution
Saponin wash buffer (see recipe) supplemented with:
50 μg/ml propidium iodide (50 μl of 1 mg/ml PI stock per 1 ml)
100 μg/ml RNase (10 μl of 10 mg/ml RNase stock per 1ml)
2 mM MgCl
2
(2 μl of 1 M MgCl
2
stock per 1 ml)
Prepare freshly and keep in the dark at 4°C before use.
Hoechst/PY staining solution
FACS buffer (see recipe) supplemented with:
2 μg/ml Hoechst 33342 (2 μl of 1 mg/ml Hoechst 33342 stock per 1 ml)
4 μg/ml Pyronin Y (4 μl of 1 mg/ml Pyronin Y stock per 1 ml)
Prepare freshly and keep in the dark at 4°C before use. Pyronin Y concentration may vary
(see Critical Parameters and Troubleshooting)
COMMENTARY
Background Information
Flow cytometric analysis of Ki-67 was described to determine the growth fraction of
lymphoma cell lines (Schwarting et al., 1986) and further applied to a cell cycle and cell
proliferation analysis on many cancer cells and hematopoietic stem cells. Pyronin Y was
first synthesized in 1889 and it has been used as a convenient histological/cytochemical dye
to stain RNA in combination with methyl green (for DNA staining). Later, double staining
of Pyronin Y and Hoechst 33342 was developed for flow cytometric analysis to estimate
DNA and RNA content in intact cells (Shapiro, 1981). These methods have been widely
used for analyzing cell cycle status.
Along with these techniques, the kinetics of cell cycle status can be assessed by cell
proliferation assays based on measuring newly-synthesized DNA content and cellular
metabolism parameters. For flow cytometric analysis of cell proliferation, genomic DNA in
replicating cells can be labeled by exposing cells to thymidine analog, 5'-bromo-2'-
deoxyuridine (BrdU) during the S phase of cell cycle. Incorporated BrdU is further stained
with fluoresceinated anti-BrdU antibodies and fluorescent DNA dye (e.g., propidium iodide,
PI; 7-Aminoactinomycin D, 7-AAD) to separate the cells according to the cell cycle of each
Kim and Sederstrom
Page 9
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

phase (i.e., G1, S, G2/M phases) (Rothaeusler and Baumgarth, 2007). A disadvantage of
BrdU incorporation method is that both membrane permeabilization and harsh DNA
denaturation processes are required for antibody penetration to the incorporated BrdU. As an
alternative of BrdU, 5-ethynyl-2'-deoxyuridine (EdU) has been developed to overcome the
limitations of BrdU method (Cappella et al., 2008; Cavanagh et al., 2011; Salic and
Mitchison, 2008). After EdU treatment during cell proliferation, incorporation of EdU can
be subsequently detected by a fluorescent azide molecule through a copper (I) catalyzed
reaction which results in a stable triazole ring formation between EdU and fluorescent dye
(called "Click reaction"). Since the small-sized fluorescent dye readily penetrates the cell
and it easily reacts with EdU even in intact DNA double strand, EdU method is highly
sensitive and much faster than a classical BrdU incorporation method. Also, EdU
incorporation assay can be combined with multiplex cell surface/intracellular staining,
which is very useful for many applications (Cappella et al., 2008; Diermeier-Daucher and
Brockhoff, 2010). The original version of the Click reaction cannot be used for multiplex
detection of some fluorophores such as GFP and R-PE which are easily damaged by high
concentration of copper and reactive oxygen species. Recently, chemical modification of
Click reaction enables to preserve GFP and R-PE fluorescence and to obtain a bright EdU
signal. This is extended to cover at least three different fluorophores (Alexa Fluor® 488,
Alexa Fluor® 647 and Pacific Blue™, see the INTERNET RESOURCES below).
Dye dilution assays using membrane-permeable fluorescent dyes are currently used to assess
cell proliferation as well. Carboxyfluorescein succinimidyl ester (CFSE or CFDA-SE,
carboxyfluorescein diacetate succinimidyl ester) is one of the widely-used fluorescent dyes
that enters the cytoplasm and covalently couples to intracellular amino acids (Lyons, 2000;
Lyons et al., 2013). Because this reaction results in extremely long-term retention of
fluorescent dye within the original cell, it was originally used to track immune cells.
Assuming that cells have homogenous cell size and undergo symmetric division, each
daughter cell has half of the parental cell volume and cellular components, as well as labeled
CFSE dyes. Thus, CFSE labeling can be applied to estimate the number of the generation
after rapid cell proliferations. Usually, CFSE dye may be traced through 6-8 generations by
flow cytometry. Similar to CFSE dye, other fluorophores for dye dilution proliferation assay
have been developed for encompassing broad range of excitation/emission spectrum (Table
2). These fluorescent dyes are better suited for multicolor applications where GFP
derivatives or FITC or similar fluorescent-conjugated antibody is used. Further, some of
dilution dyes emit in channels where cells have less natural autofluorescence that can
detected up to 10 generations during cell proliferation.
Critical Parameters
Proper cell density—Cell density should be optimized because confluent cultures may
cause growth arrest by contact inhibition, which leads to G0/G1 arrest of the cell cycle.
Excessive confluency also affects nutrient availability as well as media acidity, which may
distort experimental results. Generally, cells are harvested during the time window of
exponential growth (usually 50~70% confluency).
Kim and Sederstrom
Page 10
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Titration of antibody and fluorescent DNA dye concentration—For the first use,
antibodies against Ki-67 or surface antigen need to be titrated (usually 1:500-1:50 for flow
cytometry) to maximize the detection of signal-positive populations. Fluorescent DNA dye
concentrations may vary among cell lines and/or specific conditions, which have to be
determined empirically.
Determination of Pyronin Y concentration—Proper PY concentration is critical. Low
PY concentration does not ensure stoichiometric correlation between actual RNA level and
PY fluorescence. High PY concentration also results in condensation (precipitation) of PY-
nucleic acid complex, which interferes with PY fluorescence. Thus, optimal PY
concentration will vary among cell line, cell density and condition. Generally, titration is
required for the first use (starting from 1 μg/ml to 5 μg/ml).
Fluorescent protein analysis—To utilize BASIC PROTOCOL 1 or 2 in combination
with fluorescent proteins such as GFP and RFP, it is the best to use the ALTERNATIVE
PROTOCOL 1 as the PFA fixation followed by saponin permeabilization helps retain
structure of these proteins. Changes to BASIC PROTOCOL 1 may need to be made as GFP
and FITC spectra overlap. So, consider another non-overlapping probe for detecting Ki-67.
Some RFP derivatives also overlap with PI and, in some cases, alternative DNA probes
should be utilized.
Troubleshooting
Poor positive signal or high background fluorescence—Check appropriate laser
and filter combinations. Adjust concentration and incubation time of antibodies and
fluorescent DNA/RNA dyes. For high background fluorescence, increase FBS/BSA
concentration in the FACS buffer. In some instances (e.g., ALTERNATIVE PROTOCOL
1), PFA concentrations and incubation times may need to be adjusted to reduce background
signals. In cases where the signal is poor or non-existent with regard to surface staining,
check the manufacturer's instructions if the conjugated antibody is fixation sensitive (e.g.,
prolonged exposure to paraformaldehyde affects emission spectra of some fluorophores such
as APC-Cy™7, PE-Cy™7).
Cell clumping and extensive cell loss during fixation/washing process—
Improper fixation procedure may result in cell clumping and significant cell loss. To avoid
this, inject the cell suspension directly into the cold ethanol using a Pasteur pipette and mix
well immediately. Alternatively, use non-alcohol fixatives such as 4% paraformaldehyde
(see ALTERNATIVE PROTOCOL 1). The stained sample should be passed through a cell
strainer before analysis.
High Coefficient of Variation (CV) or wide peaks for DNA cell cycle probes—
Ensure that the samples are run in the lowest sample pressure setting possible to allow for
best interrogation of sample. Acquiring the sample in the linear setting/range of the flow
cytometer is also important. Additionally, proper cell and dye concentration is critical for
consistent histograms giving better CVs and decreasing variation between samples.
Kim and Sederstrom
Page 11
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Anticipated Results
On the basis of differences in Ki-67 expression level (Figure 1A) and RNA content (Figure
1B) of G0 cells, Basic Protocol 1 and 2 allow discrimination of resting/quiescent (G0)
population from other proliferating cells (G1, S, G2/M phases). Generally, the G0 cells have
lower levels of Ki-67 and RNA levels, so these cells may be distinguishable from other
proliferating cells. To quantify cell cycle distribution of proliferating cells precisely, fitting
software such as ModFit LT (Verity Software) and MultiCycle AV (Pheonix Flow Systems)
can be used (Darzynkiewicz and Huang, 2004).
Previous studies have demonstrated that a small portion of cells showed a significant
increase of Ki-67 level in G2/M cells (Figure 1A, asterisk) (Landberg et al., 1990). These
cells were regarded as early mitotic cells but this has not yet been firmly established. To
further separate M phase cells, other markers such as Cyclins, MPM-2 and phospho-Ser10-
histone H3 (to detect M phase) need to be combined (Juan and Darzynkiewicz, 2001;
Landberg et al., 1990; Vignon et al., 2013).
Time Considerations
Preparation and staining of Ki-67 and fluorescent DNA dye staining should take 4 hours.
The staining of Pyronin Y and Hoechst 33342 will take 3 hours. Analysis the samples
through flow cytometry will take from 1 to 10 minutes per sample depending on
concentrations. Total time is dependent upon total sample numbers.
Acknowledgement
This work was supported by the NIH (NIDDK R01DK46546) and the Cytometry and Cell Sorting Core at Baylor
College of Medicine with funding from the NIH (NIAID P30AI036211, NCI P30CA125123, and NCRR
S10RR024574)
LITERATURE CITED
Cappella P, Gasparri F, Pulici M, Moll J. Cell proliferation method: click chemistry based on BrdU
coupling for multiplex antibody staining. Curr Protoc Cytom. 2008 Chapter 7:Unit7 34.
Cavanagh BL, Walker T, Norazit A, Meedeniya AC. Thymidine analogues for tracking DNA
synthesis. Molecules. 2011; 16:7980–7993. [PubMed: 21921870]
Darzynkiewicz Z, Huang X. Analysis of cellular DNA content by flow cytometry. Curr Protoc
Immunol. 2004 Chapter 5:Unit 5 7.
Darzynkiewicz Z, Juan G, Srour EF. Differential staining of DNA and RNA. Curr Protoc Cytom. 2004
Chapter 7:Unit 7 3.
Diermeier-Daucher S, Brockhoff G. Dynamic proliferation assessment in flow cytometry. Curr Protoc
Cell Biol. 2010 Chapter 8:Unit 8 6 1-23.
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell
proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J
Immunol. 1984; 133:1710–1715. [PubMed: 6206131]
Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a
human nuclear antigen associated with cell proliferation. Int J Cancer. 1983; 31:13–20. [PubMed:
6339421]
Juan G, Darzynkiewicz Z. Bivariate analysis of DNA content and expression of cyclin proteins. Curr
Protoc Cytom. 2001 Chapter 7:Unit 7 9.
Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen
(PCNA)/cyclin during the cell cycle. Exp Cell Res. 1986; 166:209–219. [PubMed: 2874992]
Kim and Sederstrom
Page 12
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Landberg G, Tan EM, Roos G. Flow cytometric multiparameter analysis of proliferating cell nuclear
antigen/cyclin and Ki-67 antigen: a new view of the cell cycle. Exp Cell Res. 1990; 187:111–118.
[PubMed: 1967582]
Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE
dye dilution. J Immunol Methods. 2000; 243:147–154. [PubMed: 10986412]
Lyons AB, Blake SJ, Doherty KV. Flow cytometric analysis of cell division by dilution of CFSE and
related dyes. Curr Protoc Cytom. 2013 Chapter 9:Unit9 11.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer.
2009; 9:153–166. [PubMed: 19238148]
Rothaeusler K, Baumgarth N. Assessment of cell proliferation by 5-bromodeoxyuridine (BrdU)
labeling for multicolor flow cytometry. Curr Protoc Cytom. 2007 Chapter 7:Unit7 31.
Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo.
Proc Natl Acad Sci U S A. 2008; 105:2415–2420. [PubMed: 18272492]
Schluter C, Duchrow M, Wohlenberg C, Becker MH, Key G, Flad HD, Gerdes J. The cell
proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with
numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell
Biol. 1993; 123:513–522. [PubMed: 8227122]
Schwarting R, Gerdes J, Niehus J, Jaeschke L, Stein H. Determination of the growth fraction in cell
suspensions by flow cytometry using the monoclonal antibody Ki-67. J Immunol Methods. 1986;
90:65–70. [PubMed: 3711671]
Scott JE. On the mechanism of the methyl green-pyronin stain for nucleic acids. Histochemie. 1967;
9:30–47. [PubMed: 4171990]
Shapiro HM. Flow cytometric estimation of DNA and RNA content in intact cells stained with
Hoechst 33342 and pyronin Y. Cytometry. 1981; 2:143–150. [PubMed: 6170496]
Van Dilla MA, Trujillo TT, Mullaney PF, Coulter JR. Cell microfluorometry: a method for rapid
fluorescence measurement. Science. 1969; 163:1213–1214. [PubMed: 5812751]
Vignon C, Debeissat C, Georget MT, Bouscary D, Gyan E, Rosset P, Herault O. Flow cytometric
quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS
One. 2013:8–e68425.
Zetterberg A, Larsson O, Wiman KG. What is the restriction point? Curr Opin Cell Biol. 1995; 7:835–
842. [PubMed: 8608014]
Kim and Sederstrom Page 13
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript

Figure 1.
Analysis of cell cycle status by differential staining of Ki-67/DNA and DNA/RNA. HeLa
cells were fixed and subsequently stained with Ki-67-FITC and PI (A) or Hoechst 33342
and Pyronin Y (B) according to the BASIC PROTOCOL 1 and 2.
Kim and Sederstrom Page 14
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
Author Manuscript Author Manuscript Author Manuscript Author Manuscript
Author Manuscript Author Manuscript Author Manuscript Author Manuscript
Kim and Sederstrom Page 15
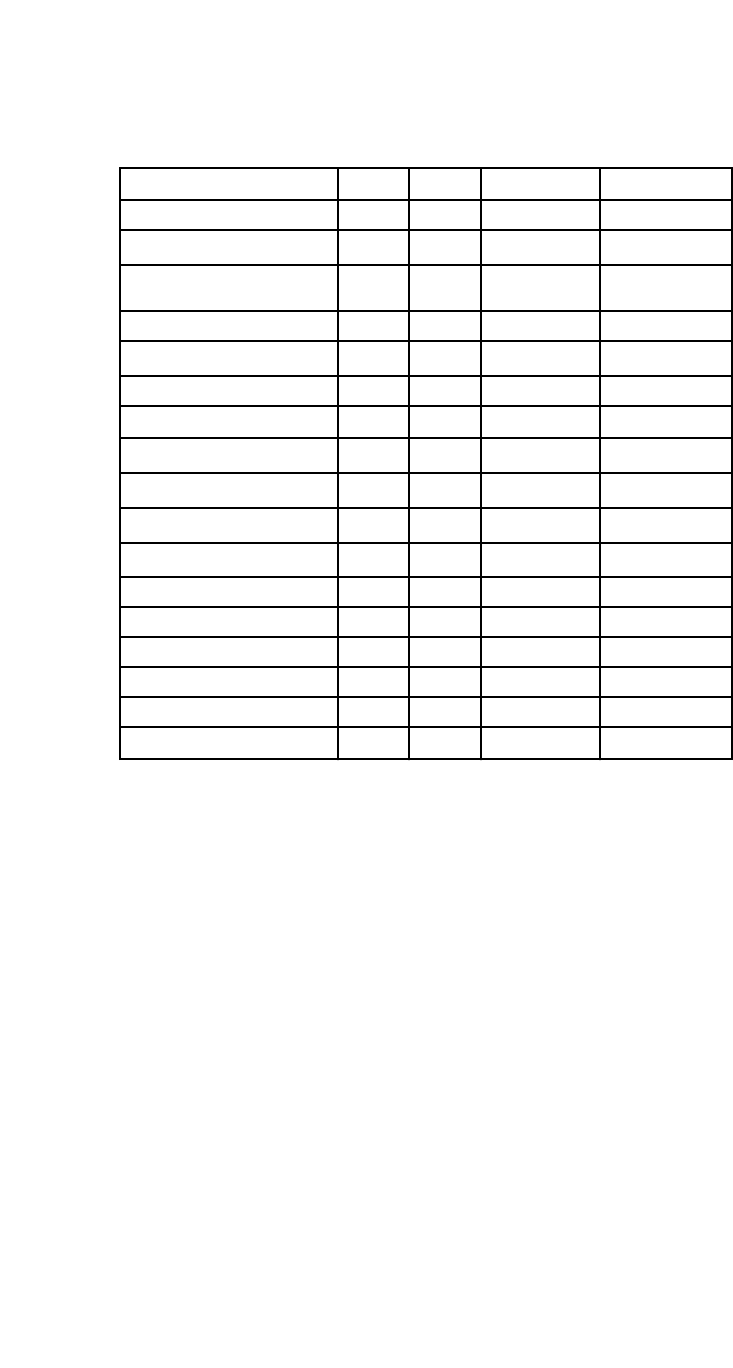
Table 1
Lists of common fluorescent DNA dyes
Commercial Name Ex
max
Em
max
Ex. Laser Manufacturer
DAPI 345 455 UV Various
Hoechst 33342
*
350 461 UV Various
Propidium Iodide (PI) 535 617
UV, 488, 532 or
561 nm
Various
7-AAD 546 647 488 or 532 nm Various
DRAQ5
*
647 681/697 633 nm Various
DRAQ7 599/644 694 633 Various
FxCycle
TM
Violet
358 461 405 nm Life Technologies
Vybrant® DyeCycle
TM
Violet
*
369 437 UV or 405 nm Life Technologies
Vybrant® DyeCycle
TM
Green
*
506 534 488 nm Life Technologies
Vybrant® DyeCycle
TM
Orange
*
519 563 488 or 532 nm Life Technologies
Vybrant® DyeCycle
TM
Ruby
*
638 686 561 or 633 nm Life Technologies
SYTOX® Blue 444 480 405 nm Life Technologies
SYTOX® Green 504 523 488 nm Life Technologies
SYTOX® Orange 547 570 488 or 532 nm Life Technologies
SYTOX® AADvanced TM 546 647 488 or 532 nm Life Technologies
SYTOX® Red 640 658 633 nm Life Technologies
FxCycle
TM
Far Red
640 658 633 nm Life Technologies
*
cell permeable dye which can stain DNA in both live or fixed cells.
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.

Author Manuscript Author Manuscript Author Manuscript Author Manuscript
Kim and Sederstrom Page 16
Table 2
Lists of fluorophores for dye dilution proliferation assay
Commercial Name Ex
max
Em
max
Ex. Laser Manufacturer
CFSE 495 519 488 nm Various
CellTrace
TM
Violet
405 450 405 nm Life Technologies
BD HorizonTM Violet Cell
Proliferation Dye (VPD450)
404 448 405 nm BD Biosciences
Cell Proliferation Dye eFluor®450 405 450 405 nm eBioscience
CytoTrack
TM
Blue
403 454 405 nm BIO-RAD
Oregon Green 488 carboxylic acid
diacetate, SE (carboxy-DFFDA SE)
496 524 488 nm Life Technologies
SNARF-1 carboxylic acid, acetate, SE 514 586 488 nm Life Technologies
CytoTrack
TM
Green
511 525 488 nm BIO-RAD
CytoTrack
TM
Yellow
542 556 532 nm BIO-RAD
CellTrace
TM
Far Red DDAO-SE
647 657 633 nm Life Technologies
Cell Proliferation Dye eFluor®670 647 670 633 nm eBioscience
CytoTrack
TM
Red
628 643 633 nm or 640 nm BIO-RAD
Curr Protoc Mol Biol. Author manuscript; available in PMC 2016 July 01.
