
Chapter 10
Coding Exon-Structure Aware Realigner (CESAR): Utilizing
Genome Alignments for Comparative Gene Annotation
Virag Sharma and Michael Hiller
Abstract
Alignment-based gene identification methods utilize sequence conservation between orthologous protein-
coding genes to annotate genes in newly sequenced genomes. CESAR is an approach that makes use of
existing genome alignments to transfer genes from one genome to other aligned genomes, and thus
generates comparative gene annotations. To accurately detect conserved exons that exhibit an intact
reading frame and consensus splice sites, CESAR produces a new alignment between orthologous exons,
taking information about the exon’s reading frame and splice site positions into account. Furthermore,
CESAR is able to detect most evolutionary splice site shifts, which helps to annotate exon boundaries at
high precision. Here, we describe how to apply CESAR to generate comparative gene annotations for one
or many species, and discuss the strengths and limitations of this approach. CESAR is available at https://
github.com/hillerlab/CESAR2.0.
Key words Comparative gene annotation, Genome alignment, CESAR, Splice site shift
1 Introduction
Identifying coding genes in genomic sequences is an important step
in annotating a genome. Several different approaches exist for this
task [1]. Transcriptome-based methods align entire or parts of
sequenced mRNAs to the genome to infer exons and introns. Ab
initio gene prediction methods detect genes solely based on char-
acteristic sequence patterns. Homology-based approaches utilize
the fact that homologous genes often have conserved sequences
and use information about genes in a related species to search for
similar sequences in the given genome.
One type of homology-based approaches makes use of align-
ments between entire genomes to project (or map) an existing gene
annotation of a “reference” species to an aligned “query” species
that lacks a gene annotation [2]. These projection approaches
assume that exons of the reference species that align well to the
query species are likely homologous exons. Thus, the coordinates
Martin Kollmar (ed.), Gene Prediction: Methods and Protocols, Methods in Molecular Biology, vol. 1962,
https://doi.org/10.1007/978-1-4939-9173-0_10, © Springer Science+Business Media, LLC, part of Springer Nature 2019
179

of aligned exon boundaries in the query genome reveal the location
of likely homologous exons (Fig. 1a).
Utilizing genome alignments for projecting gene annotations
has several advantages. First, genome alignments do not only align
exons but also the surrounding genomic context, which is helpful
to distinguish orthologs from paralogs or processed pseudogenes as
the latter are often located in a different syntenic context. Second,
many protein-coding exons are conserved over large phylogenetic
distances. If sensitive alignment parameters are used, genome align-
ments capture the majority of human coding exons in other mam-
mals and even other vertebrates [3]. Third, by making use of
existing multiple genome alignments, gene annotations can be
projected to numerous query species, as we recently demonstrated
by projecting human genes to 143 other vertebrates [3].
Despite their utility for projecting gene annotations, genome
alignments have two serious limitations. First, genome alignment
programs are not aware of the reading frame and splice sites of the
reference exon. Consequently, alignments between conserved
exons may incorrectly exhibit frameshifts or non-consensus splice
sites due to alignment ambiguities. Since one aims at projecting
only truly conserved exons that exhibit an intact reading frame and
consensus splice sites in the query species, such alignment
ccagcgcagcgggtgcggcgATGATCCTGGAGGAGAGGCCGGACGGCGCGGGCGCCGGC
gtgcgcagaactggcgcggcggcggga-----ggagg
gcgtccgagcga--gcagcgATGATCCTTGAGGAGAGGCCAGATGGCCAGGGCACTGGC
GAGGAGAGCCCGCGGCTGCAG
GAGGAAAGCTCTCGGCCGCAGgacgacggcagcatccgcaaggtgggggctgagcagg
ccagcgcagcgggtgcggcgATGATCCTGGAGGAGAGGCCGGACGGCGCGGGCGCCGGC
cagcgtccgagcgagcagcgATGATCCTTGAGGAGAGGCCAGATGGCCAGGGCACTGGC
GAGGAGAGCCCGCGGCTGCAG---------------------
GAGGAAAGCTCTCGGCCGCAGGACGACGGCAGCATCCGCAAG
coding exon (translation) start coordinates: chr15: 75704394
incorrect exon end coordinates: chr15: 75704453
correct exon end coordinates: chr15: 75704474
A
B
gtgcgcagaactggcgcggc
gt
gggggctgagcagggata
coding exon (translation) start coordinates: chr15: 75704394
genome alignment
CESAR alignment
Human
Mouse
Human
Mouse
Fig. 1 Coordinates of aligned exon boundaries do not always correspond to real exon coordi nates in another
genome. (a) Part of the genom e alignment between human and mouse that covers the first coding exon of
RHPN1 (blue font). The human consensus donor dinucl eotide (“gt”, bold) aligns to a non-consensus donor site
(red font) in the mouse, indicating that the exon end coordinates may not corre spond to the respective mouse
exon end. (b) CESAR re-aligns this sequence and detects a consensus donor site that is shifted 21 nt
downstream. These exon end coordinates precisely correspond to the exon end in mouse. Please note that the
CESAR alignment shows a 21 nt insertion in mouse. These additional 21 exonic bases are translatable in the
same reading frame
180 Virag Sharma and Michael Hiller

ambiguities cause conserved exons to be missed in the resulting
gene annotation. Second, the position of splice sites of truly con-
served exons can shift during evolution [4]. Since genome align-
ments do not aim at generating an exon alignment with consensus
splice sites, the position of the projected exon boundaries in the
query genome may be incorrect for such exons.
CESAR is a method to resolve these two limitations [4, 5]. For
a given exon, CESAR uses the query sequence provided by the
genome alignment and then re-aligns this putative exonic
sequence, incorporating both information about the reading
frame and the splice sites of the reference exon. For the given
exonic sequence, CESAR aims at finding an alignment that
(1) has consensus splice sites and (2) preserves the reading frame
and thus lacks inactivating mutations such as frameshifts and
in-frame stop codons. As a result, CESAR correctly infers exon
conservation for more than 5300 exons that had a broken reading
frame or non-consensus splice sites in the genome alignment
between human and mouse, and it is able to correctly detect
>90% of evolutionary splice site shifts [4] (Fig. 1b). This leads to
an accurate comparative gene annotation, exemplified by our obser-
vation that 99.1% of the human exons that CESAR projects to the
mouse genome overlap annotated mouse exons, and for 96.8% of
the projected exons both boundaries are correct. An example illus-
trating a gene annotation produced by CESAR is shown in Fig. 2.
We recently re-implemented CESAR in C (CESAR 2.0), which
drastically reduces runtime and memory consumption [5]. In con-
trast to the original implementation that only allowed to re-align a
single coding exon (referred to as “single-exon mode” in the
following), CESAR 2.0 also provides a “multi-exon mode” that
allows to re-align entire multi-exon genes at once against a locus in
the query genome. In multi-exon mode, CESAR 2.0 can detect
exons that do not align in the genome alignment (Fig. 3a), and it
can recognize intron deletion events that result in a larger compos-
ite exon in the query species. Furthermore, CESAR 2.0 improves
the ability to detect distal evolutionary splice site shifts, which
further enhances the precise identification of exon boundaries
[5]. In the following, we describe how to use this new implemen-
tation (simply referred to CESAR in the following) to obtain
comparative gene annotations.
2 Materials
2.1 Availability
CESAR’s source code, pre-compiled binaries, and other tools
required to annotate exons in a query genome are available from
the github repository https://github.com/hillerlab/CESAR2.0.
Open a terminal in your Linux-like environment and do the
following:
Comparative Gene Annotation with CESAR 181
git clone https://github.com/hillerlab/CESAR2.0/
cd CESAR2.0/
2.2 Installation
Compiling CESAR’s source code (written in C) requires the gcc
compiler:
make
Alternatively, a CESAR binary, pre-compiled under Linux
64 bit is present in the precompiledBinary_x86_64 subdirectory.
To automate task of annotating exons in a query genome, it is
necessary to add the “tools” subdirectory to the $PATH variable in
your environment and to set the $profilePath variable. This allows
to call the tools located in this directory without specifying the
full path.
If you are using a bash shell, do
export PATH=$PATH:‘pwd‘/tools
export profilePath=‘pwd‘
If you are using C-shell, do
setenv PATH ${PATH}:‘pwd‘/tools
setenv profilePath ‘pwd‘
2.3 Computational
Requirements
CESAR has been tested on different distributions of Linux (Cen-
tOS, Ubuntu, SUSE). The simplest way to get CESAR running is
to use the precompiled binary. For users working with a Windows
Scale 100 kb
Human Coding Exons Mapped by CESAR
Basic Gene Annotation Set from GENCODE
ASAP2
ITGB1BP1
CPSF3
IAH1
ADAM17
YWHAQ
Asap2
Itgb1bp1
Iah1
Cpsf3
Adam17
Ywhaq
Mouse genome (mm10 assembly) coordinates: chr12:21211760-21428239
Fig. 2 UCSC genom e browser screenshot showing CESAR’s gene annotation in the mouse genome. CESAR
was applied to project human exons to mouse, resulting in a gene annotation that matches the mouse
Gencode annotation. Please note that CESAR only considers coding exons. Thus, UTR exons are not projected,
as indicated by red arrows for Itgb1bp1
182 Virag Sharma and Michael Hiller

human (hg38): chr1:161,312,916-161,364,786 20 kb
Genome Alignment between Human and Mouse
SDHC
mouse (mm10): chr1:171,126,897-171,151,888
10 kb
Human Coding Exons Mapped by CESAR (single-exon mode)
S
dhc
S
DH
C
S
DH
C
Human Coding Exons Mapped by CESAR (multi-exon mode)
human (hg38): chr5:128,965,517-129,033,642
20 kb
SLC27A6
GT------------------------TATGAAGGAAGAGCAGGAATGGCTTCTATTATT
gacagCACCATTGAAAACTCTGTTTCCCAAA---------------GATATGGCATCA------
* ***** **
TAAAACCAAATACATCTTTAGATTTGGAAAAAGTTTATGAACAAGTTGTA---------CATTT
-----------------------TTGAAAAGAATGTATCACAAACTAATATTGAAACTCACTAC
*** *** * * *** * ** * ** *
CTACCAGCTTATGCTTGTCCACGATTTTTAAGAATTCAG
CTTGAG------------------------------CAAgtagt
** **
A
B
Genome Alignment between Human and Black flying-fox
TTGGTCTCTTCCCATGGCGATGTCCATCTGCCACCGTGGCACTGGTATTGCTTTGAGTGAG
tgtagATGGTCTCTTCCTATGGCACTGTCCGTTTGCCACCGAGGCTCTGGAATAGCCTTGAGTGAGgtatg
*********** ***** ***** * ******** *** **** ** ** *********
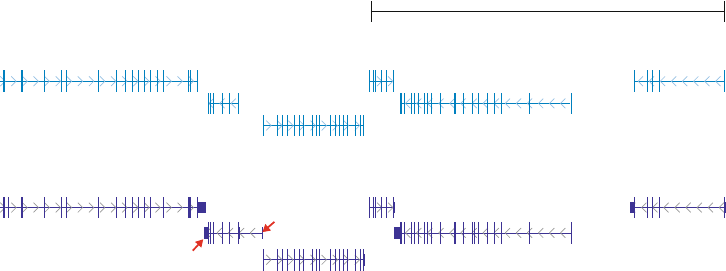
Fig. 3 Strength and weakness of CESAR’s multi-exon mode. (a) Multi-exon mode recovers an exon that does
not align in the genome alignment. Top: UCSC genome browser screenshot showing the human SDHC gene
and the genome alignment to mouse. The fourth exon does not align (red box). Bottom: UCSC genome browser
showing the orthologous mouse Sdhc gene and CESAR’s gene annotation obtained in single- and multi-exon
mode. In contrast to the single-exon mode, multi-exon mode detects exon 4 (red box) with its precise splice
sites, as shown by the sequence alignment underneath. (b) Multi-exon mode detects a false exon that is truly
absent. The ninth coding exon (red box) of human SLC27A6 does not align to the black flying-fox genome and
this coding exon is truly deleted [9]. Other exons exhibit numerous frameshifting and stop codon mutations,
showing that this gene is inactivated in the black flying-fox [9]. CESAR’s multi-exon mode neverth eless
annotates the ninth coding exon in the black flying-fox; however, the sequence alignment reveals several large
insertions and deletions and a low sequence identi ty. Thus, a post-processing step can filter out such poorly
aligning exons that are unlikely to be real
Comparative Gene Annotation with CESAR 183

machine, a virtual machine (VMware or VirtualBox) running Linux
should be able to support CESAR, though this has not been tested.
Memory requirement is proportional to the length of the
reference and query sequence. As shown in Table 1, a desktop
machine with 32 GB of RAM is sufficient to run CESAR in
single-exon mode on all human genes using the human-mouse
genome alignment. The memory requirements for CESAR’s
multi-exon mode are more demanding as intronic sequences can
be large. Still, 32 GB of RAM is sufficient to re-align 99.6% of the
human genes in their entirety to the respective mouse genomic
locus. Importantly, before allocating memory, CESAR
pre-computes an upper bound of the required memory and exits
with a warning if more memory is needed than specified by the user
with the “-maxMemory” parameter (set to 16 GB by default).
2.4 Input
CESAR’s gene-annotation workflow requires the following data as
input:
1. The genomes of the reference and all query species.
2. Transcripts annotated in the reference genome.
3. A genome alignment between the reference and one or more
query genomes.
How to obtain each input data is described below in Subhead-
ings 3.1–3.3.
Table 1
Memory requirements for CESAR for short, typical, and very long exons or genes in both single-exon
and multi-exon mode
Reference length (bp) Query length (bp) Memory (GB) Mode
100 152 0.001 Single-exon
1,005 1,170 0.01 Single-exon
5,001 4,664 0.18 Single-exon
10,227 10,038 0.77 Single-exon
984 5,484 0.04 Multi-exon
5,004 137,114 5.72 Multi-exon
9,510 135,903 10.03 Multi-exon
17,673 19,225 2.55 Multi-exon
Multi-exon mode refers to aligning all exons of the reference to the entire query locus that contains the entire
orthologous gene
184 Virag Sharma and Michael Hiller

3 Annotating Genes from a Genome Alignment
3.1 Preparing
the Genome Asse mbly
Input Data
Obtain the genome sequence of both the reference and all query
species. To this end, one can download the genome as a single file in
fasta format from NCBI (https://www.ncbi.nlm.nih.gov/assem
bly), from Ensembl (https://www.ensembl.org/downloads.html)
or from the UCSC genome browser (http://hgdownload.soe.ucsc.
edu/downloads.html). Each fasta file must be converted into a 2bit
file format by using faToTwoBit from the UCSC source code
[6]. For example, if the fasta file for mouse genome is called
“mm10.fa,” the following command converts it to a 2bit file:
faToTwoBit mm10.fa mm10.2bit
Afterward, create a “2bitDir” directory. In this directory, each
species must have a subdirectory that is identical to the assembly
name (e.g. hg38 for human, mm10 for mouse, oryAfe1 for aard-
vark). An example is provided with CESAR’s source code:
find extra/miniExample/2bitDir
which lists the following files:
extra/miniExample/2bitDir
extra/miniExample/2bitDir/hg38
extra/miniExample/2bitDir/hg38/chrom.sizes
extra/miniExample/2bitDir/hg38/hg38.2bit
extra/miniExample/2bitDir/oryAfe1
extra/miniExample/2bitDir/oryAfe1/oryAfe1.2bit
extra/miniExample/2bitDir/oryAfe1/chrom.sizes
In addition, create a file called “chrom.sizes” that contains the
size of all scaffolds for each genome by using twoBitInfo from the
UCSC source code:
for file in ‘find 2bitDir -name "*.2bit"‘ ; do
d=‘dirname $file‘;
f=‘basename $file‘;
twoBitInfo $d/$f $d/chrom.sizes;
done
3.2 Preparing
the Reference Gene
Annotation Input Data
The second step in the CESAR gene-annotation workflow is
obtaining the set of the reference species’ transcripts of which you
wish to annotate their orthologs in the query genome(s). For
example, if the reference species is human, the human Ensembl
gene annotation can be used [7]. Ensembl transcripts can be down-
loaded from Ensembl ftp site (https://www.ensembl.org/info/
Comparative Gene Annotation with CESAR 185
data/ftp/index.html) by clicking on the “GTF” link under “Gene
sets” for Human. At the time of writing, Ensembl v93 genes are
available for the human GRCh38 assembly. Clicking on “Homo_-
sapiens.GRCh38.93.gtf.gz” would save the human gene set file to
the disk. Alternatively, the UCSC genome browser provides gene
annotations, which can be downloaded in gtf format from the
Table browser (http://genome.ucsc.edu/cgi-bin/hgTables)[6].
After download, transcripts in gtf format need to be converted
to genePred format using gtfToGenePred from the UCSC source
code:
# go to the directory that contains the downloaded transcripts, e.g.
cd ~/Downloads
# unzip the file, in case it is compressed
gzip -d Homo_sapiens.GRCh38.93.gtf.gz
# this produces a file called Homo_sapiens.GRCh38.93.gtf.
# Convert to genePred format
gtfToGenePred Homo_sapiens.GRCh38.93.gtf Homo_sapiens.
GRCh38.93.gp
Ensure that the generated genePred file has the right format
(see Note 1).
Next, we filter the transcripts to retain only protein-coding
transcripts. Additionally, this filtering step also discards the follow-
ing problematic transcripts: (1) transcripts with a CDS length that
is not a multiple of 3 (e.g. genes that utilize programmed ribosomal
frameshifts or exhibit a polymorphism in the reference), and
(2) transcripts with micro-introns smaller than 30 bp as such
introns often occur in incorrectly annotated transcripts.
# At this stage, it is useful to specify the input file as a variable
# (here in Bash notation)
export inputGenes=Homo_sapiens.GRCh38.93.gp
formatGenePred.pl ${inputGenes} ${inputGenes}.CESAR ${input-
Genes}.ignore
Instead of considering all available coding transcripts of a gene,
one can run the gene-annotation workflow also with the longest
transcript only. In this case, add the “-longest” flag:
formatGenePred.pl ${inputGenes} ${inputGenes}.CESAR ${input-
Genes}.ignore -longest
3.3 Preparing
the Genome Alignment
CESAR requires as input a genome alignment between the selected
reference and one or more query genomes in maf format (https://
genome.ucsc.edu/FAQ/FAQformat.html#format5). CESAR can
handle both a pairwise or a multiple genome alignment stored in
this format. Genome alignments can be downloaded from the
186 Virag Sharma and Michael Hiller
UCSC genome browser (http://hgdownload.soe.ucsc.edu/
downloads.html). Alternatively, genome alignments can be created
with the chaining and netting pipeline [8]. The entire process of
creating a pairwise genome alignment in maf format can be auto-
mated by the UCSC script doBlastzChainNet.pl, as described in
http://genomewiki.ucsc.edu/index.php/Whole_genome_align
ment_howto.
CESAR’s workflow requires that the genome alignment is
indexed by the provided mafIndex tool, which uses the chrom.
size file of the reference genome:
mafIndex ali.maf ali.bb -chromSizes=extra/miniExample/2bit-
Dir/hg38/chrom.sizes
3.4 Preparing
and Executing
the CESAR Gene
Annotation Jobs
After preparing the three types of input (genome sequences, tran-
script information and the genome alignment), the different vari-
ables that are used as inputs to the CESAR gene-annotation
workflow need to be defined.
export reference=... # the assembly name of the reference
(e.g. hg38)
export twoBitDir=... # the directory containing the genomes
and chrom.size
# files (e.g.
extra/miniExample/2bitDir)
export alignment=... # the indexed alignment file (ali.bb
above)
export querySpecies=... # a comma-separated list of the query
species that you
# want to annotate. Each query species
must be contained
# in ${alignment}.
export outputDir=... # name of the output directory that
will contain exon
# coordinates (in subdirectories). The
directory will be
# created, if it does not exist.
export resultsDir=... # name of thedirectory that will
contain the final gene
# annotation (one gene annotation file
per query species)
export maxMemory=... # maximum amount of memory in GB that
CESAR is allowed
# to allocate
export profilePath=... # path to the directory that contains
the ’extra’
# subdirectory containing CESAR’s
profiles and matrices
Comparative Gene Annotation with CESAR 187
Next, we generate the gene-annotation workflow commands
for all filtered transcripts:
for transcript in ‘cut -f1 ${inputGenes}.forCESAR‘; do
echo "annotateGenesViaCESAR.pl ${transcript} ${alignment}
${inputGenes}.forCESAR ${reference} ${querySpecies} ${output-
Dir} ${twoBitDir} ${profilePath} -maxMemory ${maxMemory}"
done > jobList
The result is a file called “jobList” in which each line consists of
a single job that re-aligns a single transcript to all query species.
Each job is completely independent of any other job. Hence, each
job can be run in parallel on a compute cluster. In the absence of a
compute cluster, the jobs can be run sequentially:
chmod +x jobList
./jobList
Using CESAR to project 196,259 human coding exons to
mouse takes approximately 7 h on a desktop machine using a single
core. The memory requirement will vary on the size of the input
gene (see Note 2 and Table 1).
3.5 Merging CESAR’s
Output Into a Single
Gene Annotation File
per Species
In this step, we collect the results obtained in the previous step
(after each job has successfully finished) in a single genePred file for
each query species.
for species in ‘echo $querySpecies | sed ’s/,/ /g’‘; do
echo "bed2GenePred.pl $species $outputDir /dev/stdout | awk
’{if ($4 != $5) print $0}’ > $resultsDir/$species.gp"
done > jobListGenePred
chmod +x jobListGenePred
./jobListGenePred
This step takes only a few minutes. The final results are in
$resultsDir (specified as a variable above) as a single genePred-
formatted file per query species. GenePred files can be converted
to gtf format using genePredToGtf from the UCSC source code:
genePredToGtf file mm10.gp mm10.gtf
The $outputDir directory that is used to store temporary
results may be deleted afterward.
3.6 Visualizing
the Gene Annotations
This step is optional. An obtained genePred file can be visualized in
the UCSC genome browser of the query genome, as shown in
Fig. 2. This can be done by converting the genePred file into gtf
format, as described above, and then uploading this file to the
UCSC genome browser via their “Custom Track” feature.
188 Virag Sharma and Michael Hiller

3.7 Running CESAR
in Multi-exon Mode
To run CESAR in multi-exon mode, all the steps described above
are exactly the same (Subheadings 3.1–3.3 and 3.5) except Sub-
heading 3.4. After specifying the variables, the following command
will generate the jobs that run CESAR in multi-exon mode:
for transcript in ‘cut -f1 ${inputGenes}.forCESAR‘; do
echo "annotateGenesViaCESAR_multi_exon.pl ${transcript}
${alignment} ${inputGenes}.forCESAR ${reference} ${querySpe-
cies} ${outputDir} ${twoBitDir} ${profilePath} -maxMemory
${maxMemory}"
done > jobList
The jobs listed in “jobList” can be executed on a compute
cluster or run sequentially.
4 Notes
1. In case of problems with the transcript file, one can use UCSC’s
genePredCheck tool to check if the converted genePred has a
valid format.
2. A limitation of CESAR is that its memory requirement is
proportional to the lengths of the input sequences. By default,
CESAR stops with a warning if it estimates that more than
16 GB of memory may be required:
CRITICAL src/Cesar.c:117 main(): The memory consumption is
limited to 16.0000 GB by default. Your attempt requires
30.1539 GB. You can change the limit via --max-memory.
If your computer provides more memory, set the max-
Memory above to a higher value. For example, 32 GB of
RAM are sufficient to align all human exons in single-
exon mode.
In multi-exon mode, CESAR may require even more mem-
ory in case the transcript has many exons or introns in the query
genome are large. For such genes, CESAR can be run in single-
exon mode.
5 Special Cases
1. In case exons are truly deleted or overlap an assembly gap in the
query, CESAR’s multi-exon mode has a tendency to align
random intronic sequence to such reference exons, instead of
producing an alignment where these exons are entirely deleted.
Such exon alignments are characterized by large insertions and
Comparative Gene Annotation with CESAR 189
deletions and a low sequence identity (Fig. 3b). A subsequent
filtering step can be used to remove those exons from the
resulting gene annotation that are poorly aligning and thus
unlikely to be real exons.
2. CESAR’s multi-exon mode requires that all aligning exons of a
gene are located on a single locus in the query genome (same
scaffold and same strand in a co-linear order). It is therefore
recommended to use the single-exon mode for query assem-
blies with a high degree of fragmentation, where many genes
will partially align to different scaffolds. Alternatively, CESAR
can be run in both single- and multi-exon mode, and the
resulting annotations can be combined.
3. CESAR’s source code provides splice site profiles obtained for
human. These profiles are used in the re-alignment process to
locate orthologous or shifted splice sites. Splice site profiles will
be similar for closely related species such as mammals; however,
they may differ if species distantly related to human are used as
the reference. In this case, it is recommended to obtain new
splice site profiles for the reference species, which can be done
as follows:
# obtain a file that contains the longest transcript per gene
for the reference
formatGenePred.pl ${inputGenes} ${inputGenes}.CESAR
${inputGenes}.ignore -longest
# define the following variables
export input=${inputGenes}.CESAR
export ref_2bit=... # the path to the two bit file of
reference species
# extract the sequences around the splice sites from all
transcripts
extract_splic e_sit es.pl $input acc_se qs.txt donor_ seqs.t xt
$ref_2bit
# extract the sequence upstream of the first exon from the
genes
get_start_context.pl $input start_seqs.txt $ref_2bit
# Lastly, convert these sequences to profiles:
create_profiles.pl acc_seqs.txt acc_profile.txt
create_profiles.pl donor_seqs.txt do_profile.txt
create_profiles.pl start_seqs.txt firstCodon_profile.txt
# clean-up
rm acc_seqs.txt donor_seqs.txt start_seqs.txt
# Move these files to the relevant clade so that CESAR can
read these profiles
export clade=... # name of the new clade, for example chicken
mkdir -p CESAR2.0/extra/tables/$clade
190 Virag Sharma and Michael Hiller

mv acc_profile.txt CESAR2.0/extra/tables/$clade
mv do_profile.txt CESAR2.0/extra/tables/$clade
mv firstCodon_profile.txt CESAR2.0/extra/tables/$clade
# copy the original stop codon profile and the codon
substitution matrix
cp CESAR2.0/extra/tables/human/lastCodon_profile.txt
CESAR2.0/extra/tables/$clade
cp CESAR2.0/extra/tables/human/eth_codon_sub.txt
CESAR2.0/extra/tables/$clade
Acknowledgment
This work was supported by the Max Planck Society and the
German Research Foundation (HI 1423/3-1).
References
1. Picardi E, Pesole G (2010) Computational
methods for ab initio and comparative gene
finding. Methods Mol Biol 609:269–284.
https://doi.org/10.1007/978-1-60327-241-
4_16
2. Zhu J, Sanborn JZ, Diekhans M, Lowe CB,
Pringle TH, Haussler D (2007) Comparative
genomics search for losses of long-established
genes on the human lineage. PLoS Comput
Biol 3(12):e247. https://doi.org/10.1371/
journal.pcbi.0030247
3. Sharma V, Hiller M (2017) Increased alignment
sensitivity improves the usage of genome align-
ments for comparative gene annotation. Nucleic
Acids Res 45(14):8369–8377. https://doi.org/
10.1093/nar/gkx554
4. Sharma V, Elghafari A, Hiller M (2016) Coding
exon-structure aware realigner (CESAR) utilizes
genome alignments for accurate comparative
gene annotation. Nucleic Acids Res 44(11):
e103. https://doi.org/10.1093/nar/gkw210
5. Sharma V, Schwede P, Hiller M (2017) CESAR
2.0 substantially improves speed and accuracy of
comparative gene annotation. Bioinformatics 33
(24):3985–3987. https://doi.org/10.1093/
bioinformatics/btx527
6. Casper J, Zweig AS, Villarreal C, Tyner C, Speir
ML, Rosenbloom KR, Raney BJ, Lee CM, Lee
BT, Karolchik D, Hinrichs AS, Haeussler M,
Guruvadoo L, Navarro Gonzalez J, Gibson D,
Fiddes IT, Eisenhart C, Diekhans M,
Clawson H, Barber GP, Armstrong J,
Haussler D, Kuhn RM, Kent WJ (2018) The
UCSC Genome Browser database: 2018 update.
Nucleic Acids Res 46(D1):D762–D769.
https://doi.org/10.1093/nar/gkx1020
7. Zerbino DR, Achuthan P, Akanni W, Amode
MR, Barrell D, Bhai J, Billis K, Cummins C,
Gall A, Giron CG, Gil L, Gordon L,
Haggerty L, Haskell E, Hourlier T, Izuogu
OG, Janacek SH, Juettemann T, To JK, Laird
MR, Lavidas I, Liu Z, Loveland JE, Maurel T,
McLaren W, Moore B, Mudge J, Murphy DN,
Newman V, Nuhn M, Ogeh D, Ong CK,
Parker A, Patricio M, Riat HS,
Schuilenburg H, Sheppard D, Sparrow H,
Taylor K, Thormann A, Vullo A, Walts B,
Zadissa A, Frankish A, Hunt SE, Kostadima M,
Langridge N, Martin FJ, Muffato M, Perry E,
Ruffier M, Staines DM, Trevanion SJ, Aken BL,
Cunningham F, Yates A, Flicek P (2018)
Ensembl 2018. Nucleic Acids Res 46(D1):
D754–D761. https://doi.org/10.1093/nar/
gkx1098
8. Kent WJ, Baertsch R, Hinrichs A, Miller W,
Haussler D (2003) Evolution’s cauldron: dupli-
cation, deletion, and rearrangement in the
mouse and human genomes. Proc Natl Acad
Sci U S A 100(20):11484–11489. https://doi.
org/10.1073/pnas.1932072100
9. Sharma V, Hecker N, Roscito JG, Foerster L,
Langer BE, Hiller M (2018) A genomics
approach reveals insights into the importance
of gene losses for mammalian adaptations. Nat
Commun 9(1):1215. https://doi.org/10.
1038/s41467-018-03667-1
Comparative Gene Annotation with CESAR 191
