
Illumina DRAGEN Bio-IT Platform 3.7
User Guide
Document # 1000000141465 v00
October 2020
ILLUMINA PROPRIETARY
For Research Use Only. Not for use in diagnostic procedures.
This document and its contents are proprietary to Illumina, Inc. and its affiliates ("Illumina"), and are intended solely
for the contractual use of its customer in connection with the use of the product(s) described herein and for no other
purpose. This document and its contents shall not be used or distributed for any other purpose and/or otherwise
communicated, disclosed, or reproduced in any way whatsoever without the prior written consent of Illumina. Illumina
does not convey any license under its patent, trademark, copyright, or common-law rights nor similar rights of any
third parties by this document.
The instructions in this document must be strictly and explicitly followed by qualified and properly trained personnel
in order to ensure the proper and safe use of the product(s) described herein. All of the contents of this document
must be fully read and understood prior to using such product(s).
FAILURE TO COMPLETELY READ AND EXPLICITLY FOLLOW ALL OF THE INSTRUCTIONS CONTAINED HEREIN MAY
RESULT IN DAMAGE TO THE PRODUCT(S), INJURY TO PERSONS, INCLUDING TO USERS OR OTHERS, AND DAMAGE
TO OTHER PROPERTY, AND WILL VOID ANY WARRANTY APPLICABLE TO THE PRODUCT(S).
ILLUMINA DOES NOT ASSUME ANY LIABILITY ARISING OUT OF THE IMPROPER USE OF THE PRODUCT(S)
DESCRIBED HEREIN (INCLUDING PARTS THEREOF OR SOFTWARE).
© 2020 Illumina, Inc. All rights reserved.
All trademarks are the property of Illumina, Inc. or their respective owners. For specific trademark information, see
www.illumina.com/company/legal.html.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
ii
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Revision History
Document Date Description of Change
Document #
1000000141465
October
2020
Adding information on the following features:
• Single-cell RNA
• Systematic noise filtering
• DRAGEN Graph Mapper
• Forced genotyping for SV calling
• Exome auto-detect for SV calling
• BCL Conversion
• Input streaming
• CYP2D6 options
• RNA sorting and duplicate marking
• CNV Coverage Uniformity
Added the following information:
• CNV metrics file
• Tumor-normal pairs input for Somatic mode
• Joint detection in small variant calling
• Sequencing input recommendations and quality
checks for SV calling.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
iii
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Table of Contents
Revision History iii
Getting Started 1
DRAGEN Bio-IT Platform Overview 1
Software Installation 3
System Updates 4
License Usage 4
Running the System Check 5
Running Your Own Test 6
Generate a Reference 6
Prepare a Reference Genome 8
Determine Input and Output File Locations 22
Process Your Input Data 22
Example Commands for Processing FASTQ Data 22
Troubleshooting 45
Additional Resources and Support 46
DRAGEN Host Software 47
Command-line Options 47
Input Options 52
Autogenerated MD5SUM for BAM and CRAM Output Files 60
Configuration Files 61
DRAGEN DNA Pipeline 62
DNA Mapping 62
DNAAligning 65
DRAGEN Graph Mapper 72
Read Trimming 73
DRAGEN FastQC 75
ALT-Aware Mapping 78
Sorting 79
Duplicate Marking 79
Small Variant Calling 81
Copy Number Variant Calling 119
Multisample CNV Calling 142
Somatic CNV Calling 146
Repeat Expansion Detection with Expansion Hunter 152
Spinal Muscular Atrophy Calling 155
CYP2D6 Caller 156

Structural Variant Calling 160
Structural Variant De Novo Quality Scoring 176
Ploidy Calling 177
QC Metrics and Coverage/Callability Reports 183
Virtual Long Read Detection 204
Unique Molecular Identifiers 206
DRAGEN RNA Pipeline 214
Input Files 214
RNA Alignment 216
Alignment Output 216
RNA Alignment Options 219
Duplicate Marking 221
MAPQ Scoring 221
Gene Fusion Detection 221
Gene Expression Quantification 223
DRAGEN Single-Cell RNA Pipeline 223
DRAGEN Methylation Pipeline 231
DRAGEN Methylation Calling 232
Methylation-Related BAM Tags 234
Methylation Cytosine and M-Bias Reports 235
Using Bismark for Methylation Calling 235
Using TAPS Support 236
Sort and Duplicate Reads Options 236
Tools and Utilities 238
DRAGEN BCL Data Conversion 238
Monitoring System Health 247
Nirvana (Variant Annotator) 250
Hardware-Accelerated Compression and Decompression 264
Usage Reporting 264
Troubleshooting 266
How to Determine if the System is Hanging 266
Sending Diagnostic Information to Illumina Support 266
Resetting Your System after a Crash or Hang 266
Command Line Option Reference 268
General Software Options 268
Mapper Options 278
Aligner Options 278
Variant Caller Options 282
CNV Caller Options 290
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
v
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Systematic Noise Creation Options 295
Structural Variant Caller Options 295
CYP2D6_CommandLine_fDG 296
Repeat Expansion Detection Options 297
RNA-Seq Command Line Options 297
UMI Options 298
Technical Assistance 300
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
vi
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Getting Started
Before you begin, make sure that the Illumina
®
DRAGEN™ Bio-IT Platform server is turned on and that
you are logged in.
This Getting Started section helps you to start processing data as quickly as possible. It provides
instructions for the following:
DRAGEN provides tests you can run to make sure that your DRAGEN system is properly installed and
configured.Before running the tests, make sure that the DRAGEN server has adequate power and
cooling, and is connected to a network that is fast enough to move your data to and from the machine
with adequate performance.
DRAGEN Bio-IT Platform Overview
The Illumina DRAGEN™ Bio-IT Platform is based on the highly reconfigurable DRAGEN Bio-IT Processor,
which is integrated on a Field Programmable Gate Array (FPGA) card and is available in a preconfigured
server that can be seamlessly integrated into bioinformatics workflows. The platform can be loaded
with highly optimized algorithms for many different NGS secondary analysis pipelines, including the
following:
• Whole genome
• Exome
• RNA-Seq
• Methylome
• Cancer
All interaction is accomplished via DRAGEN software that runs on the host server and manages all
communication with the DRAGEN board.
This user guide summarizes the technical aspects of the system and provides detailed information for
all DRAGEN command line options.
If you are working with DRAGEN for the first time, Illumina recommends that you first read the Getting
Started on page 1 section, which provides a short introduction to DRAGEN, including running a test of
the server, generating a reference genome, and running example commands.
Input Requirements
The following are the supported input specifications for DRAGEN.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
1
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Specifications Requirement
Supported Input Files cBCL, FASTQ, BAM,
CRAM, GVCF
Upper Limit 300x coverage of
human Whole
Genome sequencing
data.
200x/100x coverage
of human T/N.
DRAGEN DNA Pipeline
Figure 1 DRAGEN DNAPipeline
The DRAGEN DNA Pipeline accelerates the secondary analysis of NGS data. For example, the time
taken to process an entire human genome at 30x coverage is reduced from approximately 10 hours
(using the current industry standard, BWA-MEM+GATK-HC software) to approximately 20 minutes.
Time scales linearly with coverage depth.
These pipelines harness the tremendous power of the DRAGEN Bio-IT Platform and include highly
optimized algorithms for mapping, aligning, sorting, duplicate marking, and haplotype variant calling.
They also use platform features such as hardware-accelerated compression and optimized BCL
conversion, together with the full set of platform tools.
Unlike all other secondary analysis methods, DRAGEN DNA Applications do not reduce accuracy to
achieve speed improvements. Accuracy for both SNPs and INDELs is improved over that of BWA-
MEM+GATK-HC in side-by-side comparisons.
In addition to haplotype variant calling, the pipeline supports calling of copy number and structural
variants as well as detection of repeat expansions.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
2
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN RNA Pipeline
DRAGEN includes an RNA-seq (splicing-aware) aligner, as well as RNA-specific analysis components
for gene expression quantification and gene fusion detection.
The DRAGEN RNA Pipeline shares many components with the DNA Pipeline. Mapping of short seed
sequences from RNA-Seq reads is performed similarly to mapping DNA reads. In addition, splice
junctions (the joining of noncontiguous exons in RNA transcripts) near the mapped seeds are detected
and incorporated into the full read alignments.
DRAGEN Methylation Pipeline
The DRAGEN Methylation Pipeline provides support for automating the processing of bisulfite
sequencing data to generate a BAM with the tags required for methylation analysis and reports
detailing the locations with methylated cytosines.
Software Installation
If you are already running the latest version of the DRAGEN software and hardware, you can skip
ahead to Run the Self Test on page 1.
You can query the current version of software and hardware with the following command:
dragen_info -b
You can query only the software version with the following command:
dragen --version
To install a new version of software or hardware, first download the package from the DRAGEN Bio-IT
Platform support pages on the Illumina website to your DRAGEN server. The preferred installation
method is to use the self-extracting .run file, as follows:
sudo sh dragen-3.3.7.el7.x86_64.run
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
3
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
During installation, if you are prompted to switch to a new hardware version, enter ‘y’. It is important
that the hardware upgrade process is not interrupted. When it is complete, you must halt and power
cycle the server. A reboot command does not update the hardware version. You must use the following
halt command to power the server off and on:
sudo ipmitool chassis power cycle
DRAGEN periodically checks for license renewal by communicating with the license server at
lus.edicogenome.com. For servers that are behind a firewall, a proxy can be configured by the network
administrator in /etc/environment. For example:
http_proxy="http://proxy.customer.com:80/"
https_proxy="https://proxy.customer.com:80/"
ftp_proxy="http://proxy.customer.com:80/"
rsync_proxy="http://proxy.customer.com:80/"
no_proxy="localhost,127.0.0.1,localaddress,.localdomain.com,.customer.com"
System Updates
DRAGEN is a flexible and extensible platform that is highly reconfigurable. Your DRAGEN subscription
allows you to download updates to the DRAGEN processors and software. These updates provide
speed, performance, throughput, and accuracy improvements.
License Usage
To check current license usage and expiration date, use the following command:
dragen_lic –f genome
The license information is output, as follows:
LICENSE_MSG| ---- Board #0 (1234565) ----
LICENSE_MSG| License Genome: used 1000.0/100000 Gbases since 2019-Jan-01
(1000000000000 bases, 1.0%)
LICENSE_MSG| Issued=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01,
period=12 months
LICENSE_MSG| -- License dongle
LICENSE_MSG| STATUS : OK
LICENSE_MSG| DONGLE SN: 0012345678900
LICENSE_MSG| RELEASE : 2016.07p5-19358
LICENSE_MSG| CHIPID : 001234567890EAD
LICENSE_MSG| DNA: active, accelerators=DNA
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| RNA: active, accelerators=RNA
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| GZIP: active, accelerators=GZIP
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| GUNZ: active, accelerators=GUNZ
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
4
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| HMM: active, accelerators=HMM
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| SMW: active, accelerators=SMW
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| RANS: active, accelerators=RANS
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
LICENSE_MSG| GRAPH: active, accelerators=GRAPH
LICENSE_MSG| issue=2019-Jan-01, start=2019-Jan-01, expiry=2020-Jan-01
The above license output example is for the Genome license. The first line shows that 1000 gigabases
have been consumed. The license installed is for 100000 gigabases and 1% of the gigabases been
used. The second line shows the license data and it is the expiry date that is important. The licenses
expires either at the expiry date or when 100% of the licensed gigabases are consumed.
Following the license data is the license information that is stored on the dongle or USB key attached to
the server. These lines show the status of all the accelerators that are enabled and they are specific to
the different pipelines. The accelerators also have an expiry date which should be similar to the license
for each pipelines and similar to the genome license example.
To obtain a new license, contact your customer service representative at
customerservice@illumina.com. If you encounter problems using your license, contact Illumina
Technical Support at techsupport@illumina.com.
Running the System Check
After turning on the server, you can make sure that your DRAGEN server is functioning properly by
running /opt/edico/self_test/self_test.sh, which does the following:
• Automatically indexes chromosome M from the hg19 reference genome
• Loads the reference genome and index
• Maps and aligns a set of reads
• Saves the aligned reads in a BAM file
• Asserts that the alignments exactly match the expected results
Each server ships with the test input FASTQ data for this script, which is located in /opt/edico/self_
test. The system check takes approximately 25–30 minutes.
The following example shows how to run the script and shows the output from a successful test.
[root@edico2 ~]# /opt/edico/self_test/self_test.sh
---------------------------------
test hash creating
test hash created
---------------------------------
reference loading /opt/edico/self_test/ref_data/chrM/hg19_chrM
reference loaded
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
5
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
---------------------------------
real0m0.640s
user0m0.047s
sys0m0.604s
not properly paired and unmapped input records percentages: PASS
---------------------------------
md5sum check dbam sorted: PASS
---------------------------------
SELF TEST COMPLETED
SELF TEST RESULT : PASS
If the output BAM file does not match expected results, then the last line of the above text is as follows:
SELF TEST RESULT : FAIL
If you experience a FAIL result after running this test script immediately after turning on your DRAGEN
server, contact Illumina Technical Support.
Running Your Own Test
When you are satisfied that your DRAGEN system is performing as expected, you are ready to run
some of your own data through the machine, as follows:
• Load the reference table for the reference genome
• Determine location of input and output files
• Process input data
Generate a Reference
If you do not have a reference, you can generate one by using the dragen –build-hash-table command
and passing in the location of the reference FASTA file. You can specify a set of parameters when
building your hash table (see the DRAGEN Bio-ITPlatform User Guide (1000000070494)).
For testing purposes, you can run the example shell script or the one of commands shown in the
examples in this guide. For these examples, the FASTA file is assumed to be located in
/staging/human/reference/hg19/hg19.fa. Change the path in the script or command line to the
correct directory, if needed. You must have change access to /staging/human/reference and its
subdirectories.
Run the example shell script as follows:
/opt/edico/examples/build_hash_table.sh
Or, run the dragen command as follows:
mkdir -p /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149
cd /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
6
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
dragen --build-hash-table true --ht-reference
/staging/human/reference/hg19/hg19.fa \
--output-dir /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--ht-alt-liftover /opt/edico/liftover/hg19_alt_liftover.sam
If you generate a hash table without including the --ht-alt-liftover option, an error similar to the
following may occur (depending on the .fa reference file used):
ERROR: Detected hg19 alternate contigs in reference at:
/staging/hg19fa/hg19.fa
DRAGENmap quality is significantly improved by building a reference with a
liftover file to enable ALT aware mapping. Use the --ht-alt-lifeover option
to specify a liftover file.
You may ignore this error and continue using your existing reference by
adding --ht-alt-aware-validate=false to your command line. However, DRAGEN
map quality will be significantly affected.
Generate the hash table with either the --ht-alt-liftover or the –ht alt-
aware-validate=false option to avoid the error listed above.
The dragen --build-hash-table command is multithreaded and defaults to eight threads. This
command takes approximately 15 minutes to run. You can use the --ht-num-threads option with a
value up to 32 (depending on the number of threads your server supports) to reduce the run time.
The hash table directory name lists key default option values that were used during the hash table
build. Illumina recommends following this best practice when you generate your own hash tables and
change the directory name accordingly.
If you enabled the CNV function, generating a hash table takes ~2 hours.
Generate an HG19 Reference
If you do not have a FASTA reference, you can get the hg19 FASTA files from UCSC and concatenate
them into a single hg19.fa file as follows:
mkdir /staging/hg19fa
cd /staging/hg19fa
wget
hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
tar -zxvf chromFa.tar.gz
cat chr*.fa > hg19.fa
Generate the DRAGEN hash table reference using the following commands.
mkdir /staging/hg19/
dragen --ht-reference /staging/hg19fa/hg19.fa \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
7
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-directory /staging/hg19/ --build-hash-table true \
--ht-alt-liftover /opt/edico/liftover/hg19_alt_liftover.sam
Load the Reference Genome
After the binary reference is loaded into memory on the DRAGEN board, it can be used for processing
any number of input data sets. You do not need to reload the reference unless you restart the system,
or need to use a different reference hash table.
The reference is loaded automatically the first time you process data with it. You can manually load the
reference genome onto the board by using the following shell script or command. The reference
directory in this example is /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149.
/opt/edico/examples/load_reference.sh
OR
dragen -l \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149
This command loads the binary reference genome into memory on the DRAGEN board, where it is used
for processing any number of input data sets. You do not need to reload the reference genome unless
you restart the system or need to switch to a different reference genome. It can take up to a minute to
load a reference genome.
DRAGEN checks whether the specified reference genome is already resident on the board. If it is, then
the upload of the reference genome is automatically skipped.You can force reloading of the same
reference genome using the force-load-reference (-l) command line option.
The command to load the reference genome prints the software and hardware versions to standard
output.For example:
DRAGEN Host Software Version 01.001.035.01.00.30.6682 and
Bio-IT Processor Version 0x1001036
After the reference genome has been loaded, the following message is printed to standard output:
DRAGEN finished normally
Prepare a Reference Genome
Before a reference genome can be used with DRAGEN, it must be converted from FASTA format into a
custom binary format for use with the DRAGEN hardware.The options used in this preprocessing step
offer tradeoffs between performance and mapping quality.
The DRAGEN system is shipped with reference genomes hg19 and GRCh37. Both reference genomes
are preinstalled based on recommended settings for general applications.If you find that performance
and mapping quality are adequate, there is a good chance that you can simply work with these supplied
reference genomes.Depending on your read lengths and other particular aspects of your application,
you may be able to improve mapping quality and/or performance by tuning the reference preprocessing
options.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
8
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Hash Table Background
The DRAGEN mapper extracts many overlapping seeds (subsequences or K-mers) from each read, and
looks up those seeds in a hash table residing in memory on its PCIe card, to identify locations in the
reference genome where the seeds match. Hash tables are ideal for extremely fast lookups of exact
matches.The DRAGEN hash table must be constructed from a chosen reference genome using the
dragen --build-hash-table option, which extracts many overlapping seeds from the reference genome,
populates them into records in the hash table, and saves the hash table as a binary file.
Reference Seed Interval
The size of the DRAGEN hash table is proportionate to the number of seeds populated from the
reference genome.The default is to populate a seed starting at every position in the reference
genome, ie, roughly 3 billion seeds from a human genome.This default requires at least 32 GB of
memory on the DRAGEN PCIe board.
To operate on larger, nonhuman genomes or to reduce hash table congestion, it is possible to populate
less than all reference seeds using the --ht-ref-seed-interval option to specify an average reference
interval.The default interval for 100% population is --ht-ref-seed-interval 1, and 50% population is
specified with --ht-ref-seed-interval 2. The population interval does not need to be an integer. For
example, --ht-ref-seed-interval 1.2 indicates 83.3% population, with mostly 1-base and some 2-base
intervals to achieve a 1.2 base interval on average.
Hash Table Occupancy
It is characteristic of hash tables that they are allocated a certain size, but always retain some empty
records, so they are less than 100% occupied. A healthy amount of empty space is important for quick
access to the DRAGEN hash table. Approximately 90% occupancy is a good upper bound. Empty space
is important because records are pseudo-randomly placed in the hash table, resulting in an abnormally
high number of records in some places.These congested regions can get quite large as the percentage
of empty space approaches zero, and queries by the DRAGEN mapper for some seeds can become
increasingly slow.
Hash Table / Seed Length
The hash table is populated with reference seeds of a single common length.This primary seed length
is controlled with the --ht-seed-len option, which defaults to 21.
The longest primary seed supported is 27 bases when the table is 8 GB to 31.5 GB in size.Generally,
longer seeds are better for run time performance, and shorter seeds are better for mapping quality
(success rate and accuracy).A longer seed is more likely to be unique in the reference genome,
facilitating fast mapping without needing to check many alternative locations.But a longer seed is also
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
9
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

more likely to overlap a deviation from the reference (variant or sequencing error), which prevents
successful mapping by an exact match of that seed (although another seed from the read may still
map), and there are fewer long seed positions available in each read.
Longer seeds are more appropriate for longer reads, because there are more seed positions available
to avoid deviations.
Value for
-ht-seed-len
Read Length
21 100 bp to 150 bp
17 to 19 shorter reads (36 bp)
27 250+ bp
Table 1 Seed Length Recommendations
Hash Table / Seed Extensions
Due to repetitive sequences, some seeds of any given length match many locations in the reference
genome.DRAGEN uses a unique mechanism called seed extension to successfully map such high-
frequency seeds.When the software determines that a primary seed occurs at many reference
locations, it extends the seed by some number of bases at both ends, to some greater length that is
more unique in the reference.
For example, a 21-base primary seed may be extended by 7 bases at each end to a 35-base extended
seed.A 21-base primary seed may match 100 places in the reference. But 35-base extensions of these
100 seed positions may divide into 40 groups of 1–3 identical 35-base seeds.Iterative seed extensions
are also supported, and are automatically generated when a large set of identical primary seeds
contains various subsets that are best resolved by different extension lengths.
The maximum extended seed length, by default equal to the primary seed length plus 128, can be
controlled with the --ht-max-ext-seed-len option. For example, for short reads, it is advisable to set the
maximum extended seed shorter than the read length, because extensions longer than the whole read
can never match.
It is also possible to tune how aggressively seeds are extended using the following options (advanced
usage):
• --ht-cost-coeff-seed-len
• --ht-cost-coeff-seed-freq
• --ht-cost-penalty
• --ht-cost-penalty-incr
There is a tradeoff between extension length and hit frequency.Faster mapping can be achieved using
longer seed extensions to reduce seed hit frequencies, or more accurate mapping can be achieved by
avoiding seed extensions or keeping extensions short, while tolerating the higher hit frequencies that
result.Shorter extensions can benefit mapping quality both by fitting seeds better between SNPs, and
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
10
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

by finding more candidate mapping locations at which to score alignments.The default extension
settings along with default seed frequency settings, lean aggressively toward mapping accuracy, with
relatively short seed extensions and high hit frequencies.
The defaults for the seed frequency options are as follows:
Option Default
--ht-cost-coeff-seed-len 1
--ht-cost-coeff-seed-freq 0.5
--ht-cost-penalty 0
--ht-cost-penalty-incr 0.7
--ht-max-seed-freq 16
--ht-target-seed-freq 4
Seed Frequency Limit and Target
One primary or extended seed can match multiple places in the reference genome. All such matches
are populated into the hash table, and retrieved when the DRAGEN mapper looks up a corresponding
seed extracted from a read. The multiple reference positions are then considered and compared to
generate aligned mapper output.However, dragen enforces a limit on the number of matches, or
frequency, of each seed, which is controlled with the --ht-max-seed-freq option.By default, the
frequency limit is 16.In practice, when the software encounters a seed with higher frequency, it
extends it to a sufficiently long secondary seed that the frequency of any particular extended seed
pattern falls within the limit. However, if a maximum seed extension would still exceed the limit, the
seed is rejected, and not populated into the hash table. Instead, dragen populates a single High
Frequency record.
This seed frequency limit does not tend to impact DRAGEN mapping quality notably, for two reasons.
First, because seeds are rejected only when extension fails, only extremely high-frequency primary
seeds, typically with many thousands of matches are rejected. Such seeds are not very useful for
mapping. Second, there are other seed positions to check in a given read.If another seed position is
unique enough to return one or more matches, the read can still be properly mapped. However, if all
seed positions were rejected as high frequency, often this means that the entire read matches similarly
well in many reference positions, so even if the read were mapped it would be an arbitrary choice, with
very low or zero MAPQ.
Thus, the default frequency limit of 16 for --ht-max-seed-freq works well. However, it may be
decreased or increased, up to a maximum of 256.A higher frequency limit tends to marginally increase
the number of reads mapped (especially for short reads), but commonly the additional mapped reads
have very low or zero MAPQ.This also tends to slow down DRAGEN mapping, because correspondingly
large numbers of possible mappings are occasionally considered.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
11
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

In addition to a frequency limit, a target seed frequency can be specified with --ht-target-seed-freq
option.This target frequency is used when extensions are generated for high frequency primary seeds.
Extension lengths are chosen with a preference toward extended seed frequencies near the
target.The default of 4 for --ht-target-seed-freq means that the software is biased toward generating
shorter seed extensions than necessary to map seeds uniquely.
Handling Decoy Contigs
The behavior of DRAGEN with respect to the handling of decoy contigs in the reference has changed
since version 2.6.
Starting with DRAGEN 3.x, DRAGEN’s hash table builder automatically detects the absence of the
decoy contigs from the reference and adds it to the FASTA file, prior to building the hash table. The
decoys file is found at /opt/edico/liftover/hs_decoys.fa. If the reference is missing the decoy
contigs, then the reads which map to the decoy contigs are artificially marked as unmapped in the
output BAM (because the original reference does not have the decoy contig). This results in an
artificially lower mapping rate, however, the accuracy of variant calling is improved thanks to removing
false positive caused by decoy reads.
Illumina recommends using this feature by default. However, you can to set the --htsuppress-decoys
option to true to suppress adding these decoys to the hash table.
The table below describes the difference in behavior between older DRAGEN versions (2.6 and earlier)
and DRAGEN 3.x versions with respect to the handling of decoy contigs in the hash table builder:
DRAGEN Behavior
DRAGEN 2.6 and earlier
versions
DRAGEN 3.x
Reference does not
include the decoy
contigs (eg, GRCh37)
Decoy reads mismap
elsewhere in the genome
due to the lack of contigs
in the reference.
• Artificially higher
mapping rate.
• False positive calls in
noisy regions to which
the decoy contigs are
mismapped.
DRAGEN automatically detects the
absence of the decoy contig from the
reference and adds it to the FASTA file.
• Artificially lower mapping rate
(because decoy reads which map to
the decoy contigs are artificially
marked as unmapped in the output
BAM (because the original reference
does not have the decoy contig) .
• False positive calls are avoided thanks
to adding the decoy contigs under the
hood. Therefore this helps variant
calling.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
12
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN Behavior
DRAGEN 2.6 and earlier
versions
DRAGEN 3.x
Reference includes
the decoy contigs
(eg, hs37d5)
Decoy reads map to the
decoy contigs.
• High mapping rate
• No false positive calls
caused by decoy reads
because decoy reads
map to the right place
Decoy reads map to the decoy contig.
• High mapping rate
• No false positive calls caused by decoy
reads because decoy reads map to the
right place
ALT-Aware Hash Tables
To enable ALT-aware mapping in DRAGEN, build GRch38 (and other references with ALT contigs) with
a liftover file by using the --ht-alt-liftover option.The hash table builder classifies each reference
sequence as primary or alternate based on the liftover file, and packs primaries before alternates in
reference.bin.SAM liftover files for hg38DH and hg19 are in the /opt/edico/liftover folder. The --
ht-alt-liftover option specifies the path to the liftover file to build an ALT-aware hash table.
To override the liftover file requirement, set the --ht-alt-aware-validate option to false when building
the hash tables and when running dragen.
Custom Liftover Files
Custom liftover files can be used in place of those provided with DRAGEN.Liftover files must be SAM
format, but no SAM header is required. SEQ and QUAL fields can be omitted (‘*’).Each alignment record
should have an alternate haplotype reference sequence name as QNAME, indicating the RNAME and
POS of its liftover alignment in a destination (normally primary assembly) reference sequence.
Reverse-complemented alignments are indicated by bit 0x10 in FLAG.Records flagged unmapped
(0x4) or secondary (0x100) are ignored.The CIGAR may include hard or soft clipping, leaving parts of
the ALT contig unaligned.
A single reference sequence cannot serve as both an ALT contig (appearing in QNAME) and a liftover
destination (appearing in RNAME).Multiple ALT contigs can align to the same primary assembly
location.Multiple alignments can also be provided for a single ALT contig (extras optionally be flagged
0x800 supplementary), such as to align one portion forward and another portion reverse-
complemented. However, each base of the ALT contig only receives one liftover image, according to
the first alignment record with an M CIGAR operation covering that base.
SAM records with QNAME missing from the reference genome are ignored, so that the same liftover file
may be used for various reference subsets, but an error occurs if any alignment has its QNAME present
but its RNAME absent.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
13
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Command Line Options
Use the --build-hash-table option to transform a reference FASTA into the hash table for DRAGEN
mapping. It takes as input a FASTA file (multiple reference sequences being concatenated) and a
preexisting output directory and generates the following set of files:
reference.bin
The reference sequences, encoded in 4 bits per base. Four-bit codes
are used, so the size in bytes is roughly half the reference genome
size. In between reference sequences, N are trimmed and padding is
automatically inserted.For example, hg19 has 3,137,161,264 bases in
93 sequences. This is encoded in 1,526,285,312 bytes = 1.46 GB,
where 1 GB means 1 GiB or 2
30
bytes.
hash_table.cmp
Compressed hash table.The hash table is decompressed and used by
the DRAGEN mapper to look up primary seeds with length specified by
the --ht-seed-len option and extended seeds of various lengths.
hash_table.cfg
A list of parameters and attributes for the generated hash table, in a
text format. This file provides key information about the reference
genome and hash table.
hash_
table.cfg.bin
A binary version of hash_table.cfg used to configure the DRAGEN
hardware.
hash_table_
stats.txt
A text file listing extensive internal statistics on the constructed hash
including the hash table occupancy percentages. This table is for
information purposes. It is not used by other tools.
Build command usage is as follows:
dragen --build-hash-table true [options] --ht-reference <reference.fasta> -
-output-directory <outdir>
The sections that follow provide information on the options for building a hash table.
Input/Output Options
The --ht-reference and --output-directory options are required for building a hash table.The --
ht-reference option specifies the path to the reference FASTA file, while --output-directory specifies a
preexisting directory where the hash table output files are written.Illumina recommends organizing
various hash table builds into different folders. As a best practice, folder names should include any
nondefault parameter settings used to generate the contained hash table. The sequence names in the
reference FASTA file must be unique.
Primary Seed Length
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
14
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The --ht-seed-len option specifies the initial length in nucleotides of seeds from the reference genome
to populate into the hash table.At run time, the mapper extracts seeds of this same length from each
read, and looks for exact matches (unless seed editing is enabled) in the hash table.
The maximum primary seed length is a function of hash table size. The limit is k=27 for table sizes from
16 GB to 64 GB, covering typical sizes for whole human genome, or k=26 for sizes from 4 GB to 16 GB.
The minimum primary seed length depends mainly on the reference genome size and complexity. It
needs to be long enough to resolve most reference positions uniquely. For whole human genome
references, hash table construction typically fails with k < 16. The lower bound may be smaller for
shorter genomes, or higher for less complex (more repetitive) genomes. The uniqueness threshold of -
-ht-seed-len 16 for the 3.1Gbp human genome can be understood intuitively because log4(3.1 G) ≈ 16,
so it requires at least 16 choices from 4 nucleotides to distinguish 3.1 G reference positions.
Accuracy Considerations
For read mapping to succeed, at least one primary seed must match exactly (or with a single SNP when
edited seeds are used).Shorter seeds are more likely to map successfully to the reference, because
they are less likely to overlap variants or sequencing errors, and because more of them fit in each
read.So for mapping accuracy, shorter seeds are mainly better.
However, very short seeds can sometimes reduce mapping accuracy.Very short seeds often map to
multiple reference positions, and lead the mapper to consider more false mapping locations.Due to
imperfect modeling of mutations and errors by Smith-Waterman alignment scoring and other
heuristics, occasionally these noise matches may be reported.Run time quality filters such as --
Aligner.aln_min_score can control the accuracy issues with very short seeds.
Speed Considerations
Shorter seeds tend to slow down mapping, because they map to more reference locations, resulting in
more work such as Smith-Waterman alignments to determine the best result.This effect is most
pronounced when primary seed length approaches the reference genome’s uniqueness threshold, eg,
K=16 for whole human genome.
Application Considerations
• Read Length—Generally, shorter seeds are appropriate for shorter reads, and longer seeds for
longer reads.Within a short read, a few mismatch positions (variants or sequencing errors) can
chop the read into only short segments matching the reference, so that only a short seed can fit
between the differences and match the reference exactly.For example, in a 36 bp read, just one
SNP in the middle can block seeds longer than 18 bp from matching the reference.By contrast, in a
250 bp read, it takes 15 SNPs to exceed a 0.01% chance of blocking even 27 bp seeds.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
15
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Paired Ends—The use of paired end reads can make longer seeds yield good mapping
accuracy.DRAGEN uses paired end information to improve mapping accuracy, including with
rescue scans that search the expected reference window when only one mate has seeds mapping
to a given reference region.Thus, paired end reads have essentially twice the opportunity for an
exact matching seed to find their correct alignments.
• Variant or Error Rate—When read differences from the reference are more frequent, shorter
seeds may be required to fit between the difference positions in a given read and match the
reference exactly.
• Mapping Percentage Requirement—If the application requires a high percentage of reads to be
mapped somewhere (even at low MAPQ), short seeds may be helpful.Some reads that do not
match the reference well anywhere are more likely to map using short seeds to find partial matches
to the reference.
Maximum Seed Length
The --ht-max-ext-seed-len option limits the length of extended seeds populated into the hash table.
Primary seeds (length specified by --ht-seed-len) that match many reference positions can be
extended to achieve more unique matching, which may be required to map seeds within the maximum
hit frequency (--ht-max-seed-freq).
Given a primary seed length k, the maximum seed length can be configured between k and k+128. The
default is the upper bound, k+128.
When to Limit Seed Extension
The --ht-max-ext-seed-len option is recommended for short reads, eg, less than 50 bp.In such cases,
it is helpful to limit seed extension to the read length minus a small margin, such as 1–4 bp. For
example, with 36 bp reads, setting --ht-max-ext-seed-len to 35 might be appropriate.This ensures
that the hash table builder does not plan a seed extension longer than the read causing seed extension
and mapping to fail at run time, for seeds that could have fit within the read with shorter extensions.
While seed extension can be similarly limited for longer reads, eg, setting --ht-max-ext-seed-len to 99
for 100 bp reads, there is little utility in this because seeds are extended conservatively in any event.
Even with the default k+128 limit, individual seeds are only extended to the lengths required to fit under
the maximum hit frequency (--ht-max-seed-freq), and at most a few bases longer to approach the
target hit frequency (--ht-target-seed-freq), or to avoid taking too many incremental extension steps.
Maximum Hit Frequency
The --ht-max-seed-freq option sets a firm limit on the number of seed hits (reference genome
locations) that can be populated for any primary or extended seed. If a given primary seed maps to
more reference positions than this limit, it must be extended long enough that the extended seeds
subdivide into smaller groups of identical seeds under the limit. If, even at the maximum extended seed
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
16
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
length (--ht-max-ext-seed-len), a group of identical reference seeds is larger than this limit, their
reference positions are not populated into the hash table. Instead, dragen populates a single High
Frequency record.
The maximum hit frequency can be configured from 1 to 256.However, if this value is too low, hash
table construction can fail because too many seed extensions are needed. The practical minimum for a
whole human genome reference, other options being default, is 8.
Accuracy Considerations
Generally, a higher maximum hit frequency leads to more successful mapping.There are two reasons
for this. First, a higher limit rejects fewer reference positions that cannot map under it. Second, a higher
limit allows seed extensions to be shorter, improving the odds of exact seed matching without
overlapping variants or sequencing errors.
However, as with very short seeds, allowing high hit counts can sometimes hurt mapping
accuracy.Most of the seed hits in a large group are not to the true mapping location, and occasionally
one of these noise hits may be reported due to imperfect scoring models.Also, the mapper limits the
total number of reference positions it considers, and allowing very high hit counts can potentially crowd
out the actual best match from consideration.
Speed Considerations
Higher maximum hit frequencies slow down read mapping, because seed mapping finds more
reference locations, resulting in more work, such as Smith-Waterman alignments, to determine the
best result.
ALT-Aware Liftover File Options
The following options control See ALT-Aware Hash Tables on page 13 for more information on building
a custom liftover file.
• --ht-alt-liftover
The --ht-alt-liftover option specifies the path to the liftover file to build an ALT-aware hash table.
This option is required when building from a reference with ALT contigs. SAM liftover files for
hg38DH and hg19 are provided in /opt/edico/liftover.
• --ht-alt-aware-validate
When building hash tables from a reference that contains ALT-contigs, building with a liftover file is
required. To disable this requirement, set the --ht-alt-aware-validate option to false.
• --ht-decoys
The DRAGEN software automatically detects the use of hg19 and hg38 references and adds
decoys to the hash table when they are not found in the FASTA file. Use the --ht-decoys option to
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
17
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
specify the path to a decoys file. The default is /opt/edico/liftover/hs_decoys.fa.
• --ht-suppress-decoys
Use the --ht-suppress-decoys option to suppress the use of the decoys file when building the hash
table.
Graph Mapper Hash Table Options
The following are additional ALT-aware file options to control building hash tables for the DRAGEN
graph mapper.
• --ht-pop-alt-contigs —Specifies the path to the reference FASTA file with population alternate
contigs. The standard reference FASTA is augmented with the population alternate contigs during
hash table build. The population alternate contigs file must have a corresponding liftover SAM file.
A population alternate contig file for hg38 reference is provided in /opt/edico/liftover (pop_
altContig.fa.gz).
• --ht-pop-alt-liftover—Specifies the path to the liftover file for the population alternate contigs. The
liftover SAM file must have a corresponding population alternate contigs FASTA. A population
alternate contig SAM liftover file for hg38 reference is provided in /opt/edico/liftover (pop_
liftover.sam.gz).
• --ht-pop-snps—Specifies the path to a VCF file containing unphased population SNPs. The
standard reference FASTA is augmented with these SNPs as multibase codes during mapping-
aligning. Each SNP entry in the VCF only requires the CHROM, POS, REF, ALT columns. The ALT
column can have multiple comma-separated population SNP VCF for hg38 reference is provided in
/opt/edico/liftover (pop_snps.vcf.gz).
DRAGENSoftware Options
• --ht-num-threads
The --ht-num-threads option determines the maximum number of worker CPU threads that are
used to speed up hash table construction. The default for this option is 8, with a maximum of 32
threads allowed.
If your server supports execution of more threads, it is recommended that you use the maximum.
For example, the DRAGEN servers contain 24 cores that have hyperthreading enabled, so a value
of 32 should be used. When using a higher value, adjust --ht-max-table-chunks needs to be
adjusted as well. The servers have 128 GB of memory available.
• --ht-max-table-chunks
The --ht-max-table-chunks option controls the memory footprint during hash table construction by
limiting the number of ~1 GB hash table chunks that reside in memory simultaneously. Each
additional chunk consumes roughly twice its size (~2 GB) in system memory during construction.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
18
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The hash table is divided into power-of-two independent chunks, of a fixed chunk size, X, which
depends on the hash table size, in the range 0.5 GB < X ≤ 1 GB. For example, a 24 GB hash table
contains 32 independent 0.75 GB chunks that can be constructed by parallel threads with enough
memory and a 16 GB hash table contains 16 independent 1 GB chunks.
The default is --ht-max-table-chunks equal to --ht-num-threads, but with a minimum default --ht-
max-table-chunks of 8. It makes sense to have these two options match, because building one
hash table chunk requires one chunk space in memory and one thread to work on it. Nevertheless,
there are build-speed advantages to raising --ht-max-table-chunks higher than --ht-num-threads,
or to raising --ht-num-threads higher than --ht-max-table-chunks.
Size Options
• --ht-mem-limit—Memory Limit
The --ht-mem-limit option controls the generated hash table size by specifying the DRAGEN board
memory available for both the hash table and the encoded reference genome.The --ht-mem-limit
option defaults to 32 GB when the reference genome approaches WHG size, or to a generous size
for smaller references. Normally there is little reason to override these defaults.
• --ht-size—Hash Table Size
This option specifies the hash table size to generate, rather than calculating an appropriate table
size from the reference genome size and the available memory (option --ht-mem-limit). Using
default table sizing is recommended and using --ht-mem-limit is the next best choice.
Seed Population Options
• --ht-ref-seed-interval—Seed Interval
The --ht-ref-seed-interval option defines the step size between positions of seeds in the reference
genome populated into the hash table. An interval of 1 (default) means that every seed position is
populated, 2 means 50% of positions are populated, etc. Noninteger values are supported, eg, 2.5
yields 40% populated.
Seeds from a whole human reference are easily 100% populated with 32 GB memory on DRAGEN
boards. If a substantially larger reference genome is used, change this option.
• --ht-soft-seed-freq-cap and --ht-max-dec-factor—Soft Frequency Cap and Maximum Decimation
Factor for Seed Thinning
Seed thinning is an experimental technique to improve mapping performance in high-frequency
regions.When primary seeds have higher frequency than the cap indicated by the --ht-soft-seed-
freq-cap option, only a fraction of seed positions are populated to stay under the cap.The --ht-
max-dec-factor option specifies a maximum factor by which seeds can be thinned. For example, --
ht-max-dec-factor 3 retains at least 1/3 of the original seeds. --ht-max-dec-factor 1 disables any
thinning.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
19
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Seeds are decimated in careful patterns to prevent leaving any long gaps unpopulated. The idea is
that seed thinning can achieve mapped seed coverage in high frequency reference regions where
the maximum hit frequency would otherwise have been exceeded. Seed thinning can also keep
seed extensions shorter, which is also good for successful mapping.Based on testing to date, seed
thinning has not proven to be superior to other accuracy optimization methods.
• --ht-rand-hit-hifreq and --ht-rand-hit-extend—Random Sample Hit with HIFREQ Record and
EXTENDRecord
Whenever a HIFREQ or EXTEND record is populated into the hash table, it stands in place of a large
set of reference hits for a certain seed.Optionally, the hash table builder can choose a random
representative of that set, and populate that HIT record alongside the HIFREQ or EXTEND record.
Random sample hits provide alternative alignments that are very useful in estimating MAPQ
accurately for the alignments that are reported. They are never used outside of this context for
reporting alignment positions, because that would result in biased coverage of locations that
happened to be selected during hash table construction.
To include a sample hit, set --ht-rand-hit-hifreq to 1. The --ht-rand-hit-extend option is a minimum
pre-extension hit count to include a sample hit, or zero to disable.Modifying these options is not
recommended.
Seed Extension Control
DRAGEN seed extension is dynamic, applied as needed for particular K-mers that map to too many
reference locations.Seeds are incrementally extended in steps of 2–14 bases (always even) from a
primary seed length to a fully extended length. The bases are appended symmetrically in each
extension step, determining the next extension increment if any.
There is a potentially complex seed extension tree associated with each high frequency primary seed.
Each full tree is generated during hash table construction and a path from the root is traced by iterative
extension steps during seed mapping.The hash table builder employs a dynamic programming
algorithm to search the space of all possible seed extension trees for an optimal one, using a cost
function that balances mapping accuracy and speed. The following options define that cost function:
• --ht-target-seed-freq—Target Hit Frequency
The --ht-target-seed-freq option defines the ideal number of hits per seed for which seed
extension should aim.Higher values lead to fewer and shorter final seed extensions, because
shorter seeds tend to match more reference positions.
• --ht-cost-coeff-seed-len—Cost Coefficient for Seed Length
The --ht-cost-coeff-seed-len option assigns the cost component for each base by which a seed is
extended. Additional bases are considered a cost because longer seeds risk overlapping variants or
sequencing errors and losing their correct mappings. Higher values lead to shorter final seed
extensions.
• --ht-cost-coeff-seed-freq—Cost Coefficient for Hit Frequency
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
20
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The --ht-cost-coeff-seed-freq option assigns the cost component for the difference between the
target hit frequency and the number of hits populated for a single seed. Higher values result
primarily in high-frequency seeds being extended further to bring their frequencies down toward
the target.
• --ht-cost-penalty—Cost Penalty for Seed Extension
The --ht-cost-penalty option assigns a flat cost for extending beyond the primary seed length. A
higher value results in fewer seeds being extended at all. Current testing shows that zero (0) is
appropriate for this parameter.
• --ht-cost-penalty-incr—Cost Increment for Extension Step
The --ht-cost-penalty-incr option assigns a recurring cost for each incremental seed extension
step taken from primary to final extended seed length. More steps are considered a higher cost
because extending in many small steps requires more hash table space for intermediate EXTEND
records, and takes substantially more run time to execute the extensions. A higher value results in
seed extension trees with fewer nodes, reaching from the root primary seed length to leaf
extended seed lengths in fewer, larger steps.
Pipeline Specific Hash Tables
When building a hash table, DRAGEN configures the options to work for DNA-seq processing by
default. To run RNA-Seq data, you must build an RNA-Seq hash table using the --ht-build-rna-
hashtable true option. For an RNA-Seq alignment run, refer to the original --output-directory, not to the
automatically generated subdirectory.
The CNV pipeline requires that the hash table be built with --enable-cnv set to true, which generates
an additional k-mer hashmap that is used in the CNV algorithm. Illumina recommends that that you
always use the --enable-cnv option, in case you wish to perform CNV calling with the same hash table
that is used for mapping and aligning.
DRAGEN methylation runs require building a special pair of hash tables with reference bases converted
from C->T for one table, and G->A for the other. When running the hash table generation with the --ht-
methylated option, these conversions are done automatically, and the converted hash tables are
generated in a pair of subdirectories of the target directory specified with --output-directory. The
subdirectories are named CT_converted and GA_converted, corresponding to the automatic base
conversions. When using these hash tables for methylated alignment runs, refer to the original --
output-directory and not to either of the automatically generated subdirectories.
These base conversions remove a significant amount of information from the hashtables, so you may
find it necessary to tune the hash table parameters differently than you would in a conventional hash
table build.The following options are recommended for building hash tables for mammalian species:
dragen --build-hash-table=true --output-directory $REFDIR \
--ht-reference $FASTA --ht-max-seed-freq 16 \
--ht-seed-len 27 --ht-num-threads 40 --ht-methylated=true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
21
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Determine Input and Output File Locations
The DRAGEN Bio-IT Platform is very fast, which requires careful planning for the locations of the input
and output files.If the input or output files are on a slow file system, then the overall performance of
the system is limited by the throughput of that file system. It is recommended that inputs and outputs
are streamed directly from/to a mounted external storage system.
The DRAGEN system is preconfigured with at least one fast file system consisting of a set of fast SSD
disks grouped with RAID-0 for performance.This file system is mounted at /staging. This name was
chosen to emphasize the fact that this area was built to be large and fast, but is not redundant. Failure
of any of the file system's constituent disks leads to the loss of all data stored there.
During processing, DRAGEN generates and reads back temporary files. With DRAGEN, it is highly
recommended to always direct temporary files to the fast SSD (or /staging) by using the --
intermediate-results-dir option. If the --intermediate-results-dir option is not provided, temporary files
are written to the --output-directory. DRAGEN recommends streaming inputs and outputs using
an mounted external storage system.
Process Your Input Data
To analyze FASTQ data, use the dragen command.For example, the following command can be used
to analyze a single-ended FASTQ file:
dragen \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/test/data/SRA056922.fastq \
--output-directory /staging/test/output \
--output-file-prefix SRA056922_dragen \
--RGID DRAGEN_RGID \
--RGSM DRAGEM_RGSM
For more information on the command line options, see DRAGEN Host Software on page 47
Example Commands for Processing FASTQ Data
After you have loaded your reference, you can process input FASTQ data. Choose the example that
best matches your data sets. These commands can take up to approximately 30 minutes to run on a 24
core server with SSD drives on a 30x coverage whole human genome when running end-to-end
(FASTQ input to VCF output). The speed scales with input size, so a 60x coverage genome would take
twice as long. Exome data takes a fraction of the time. A successful result is indicated by the following
message (an application exit code of 0 when run from a script):
DRAGEN finished normally
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
22
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
This message is followed by a block of metrics such as read count and performance. If there is a
problem with the command line options, an error is displayed, followed by help usage. You may need to
scroll up to see the error.
The DRAGEN log can be redirected to a file, to keep the record for future reference.
To get help on dragen command line options, run the following command:
dragen -h
The example commands shown in this document are formatted for visual display and include line feed
characters. To avoid copy-paste errors, each example command is contained in an individual shell
script in /opt/edico/examples/. These examples have the following requirements:
• All commands accept either FASTQ or gzipped FASTQ (fastq.gz). DRAGEN automatically
determines the file type.
• All commands include the -f option, which forces the output file to be overwritten if it exists.
• All commands assume that your DRAGEN reference (hash table) directory is
/staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149, and your FASTA reference file is
/staging/human/reference/hg19/hg19.fa. Replace those with the correct references or directory
paths, if needed.
• All command examples assume that the example data package is in /staging/examples (in
particular, the .fastq and fastq.gz files are expected to be in /staging/examples/reads).
• To run these example commands, you must have write access to the /staging/examples folder.
End-to-End Aligning and Variant Calling Examples
Paired-End BAM Input, VCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b /staging/human/unsorted_SRA056922_30x_e10_50M.bam \
--enable-map-align true \
--enable-map-align-output true \
--enable-variant-caller true \
--vc-sample-name Unsorted_SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true
• Or, run /opt/edico/examples/paired_fastq_in_dupmark_bam_and_vcf_out.sh.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
23
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
If the /staging/human/unsorted_SRA056922_30x_e10_50M.bam input file for the example above is
missing, run the /opt/edico/examples/paired_fastq_in_unsorted_bam_out.sh script o
generate it.
Paired-End FASTQ Input, VCF Output (Default)
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
• Or, run /opt/edico/examples/paired_fastq_in_vcf_out.sh.
This example shows the minimum options that must be specified to perform an end-to-end run. By
default, duplicate-marking is not performed and no BAM output is produced.
Paired-End Fastq Input, Sorted and Duplicate-Marked, VCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true
• Or, run /opt/edico/examples/paired_fastq_in_dupmark_vcf_out.sh.
Paired-End FASTQ Input, Sorted BAM and VCF Output
• Enter the following input:
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
24
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true \
--enable-map-align-output true
• Or, run /opt/edico/examples/sorted_bam_in_vcf_out.sh.
Paired-End FASTQ Input, Sorted SAM and VCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true \
--enable-map-align-output true \
--output-format SAM
• Or, run /opt/edico/examples/paired_fastq_in_dupmark_sam_and_vcf_out.sh.
Paired-End FASTQ Input, Sorted CRAM and VCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true \
--enable-map-align-output true \
--output-format CRAM \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
25
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Or, run /opt/edico/examples/paired_fastq_in_dupmark_cram_and_vcf_out.sh.
Paired-End FASTQ Input, Sorted BAM and VCF Output, plus Repeat Genotyping VCF
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-duplicate-marking true \
--enable-map-align-output true \
--repeat-genotype-enable true \
--repeat-genotype-specs /opt/edico/repeat-specs/hg19 \
--repeat-genotype-sex female \
--repeat-genotype-ref-fasta /staging/human/reference/h19/hg19.fa
Alignment Only Examples
All the variations for performing alignment shown in these examples can be used in the end-to-end
case as well.
Map/Align Single-Ended FASTQ Input, Sorted BAM output (Default)
• Enter the following input:
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_rand1_100K.fastq \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_rand1_100K \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM \
• Or, run /opt/edico/examples/single_fastq_in_bam_out.sh.
Map/Align Single-ended FASTQ input, Sorted, Duplicate-Marked BAM Output
• Enter the following input:
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_rand1_100K.fastq \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
26
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_rand1_100K_dup_marked \
--enable-duplicate-marking true \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/single_fastq_in_dupmark_bam_out.sh.
Map/Align Paired-End FASTQ Input, Sorted BAM Output (Default)
• Enter the following input:
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/paired_fastq_in_bam_out.sh.
Map/Align Paired-End FASTQ Input, Sorted CRAM Output
• Enter the following input:
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--output-format CRAM \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/paired_fastq_in_cram_out.sh.
Map/Align Paired-End FASTQ Input, Sorted Uncompressed BAM Output
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix uncompressed_SRA \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
27
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--enable-bam-compression false \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
Map/Align Paired-End FASTQ Input, Sorted SAM Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--output-format SAM \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/paired_fastq_in_sam_out.sh.
Map/Align Paired -End FASTQ Input, UN-Sorted BAM output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix unsorted_SRA056922_30x_e10_50M \
--enable-sort false \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/paired_fastq_in_unsorted_bam_out.sh.
Map/Align Interleaved Paired-Ended FASTQ Input, BAM Output
• Enter the following input:
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_PE_30x_rand1_10K_interleaved.fastq \
--interleaved \
--output-directory /staging/examples/ \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
28
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-file-prefix SRA056922_PE_30x_rand1_10K_interleaved \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
• Or, run /opt/edico/examples/interleaved_fastq_in_bam_out.sh.
RNA Map and Align Only Examples
Any of the Map/Align Only examples can be used for RNA. The only difference in the command is to set
the --enable-rna option to true. DRAGEN automatically picks up the RNA-specific hash tables and uses
the RNA spliced aligner in its processing.
The hash table used for these examples must be generated with the --ht-build-rna-hashtable true
option. Otherwise, the run will fail with an error similar to the following:
ERROR: The specified hashtable directory cannot be used to run RNA:
/staging/examples/reference/hg19/hg19.fa.k_21.f_16.m_149
If this error occurs, regenerate the hash table with the --ht-build-rna-hashtable true option.
RNA Map/Align Paired-Ended FASTQ Input, BAM Output
dragen –f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--enable-rna true \
--RGID DRAGEN_RGID \
--RGSM DRAGEN_RGSM
The following is example command-line to map-align RNA-seq data with additional command line
options, including a path to a gtf file with gene annotations. The gene annotations file improves
mapping by providing a list of known splice-junctions (rather than discovering them all de novo.)
dragen -f \
-r <HASHTABLE_DIR>
-1 <FASTQ1> \
-2 <FASTQ2> \
-a $/reference_genomes/annotation/GTF/$gencode.annotation.gtf
--enable-map-align true \
--enable-sort=true \
--enable-bam-indexing true \
--enable-map-align-output true \
--output-format=BAM \
--RGID=<READ_GROUP_ID> \
--RGSM=<Sample_NAME> \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
29
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

--RGPL=<LIBRARY> \
--config-file /opt/edico/config/dragen-user-defaults.cfg \
--enable-rna=true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX>
RNAQuantification
To run gene and transcript expression quantification add the following option:
--enable-rna-quantification true
When --enable-rna-quantification is set to true, GCbias correction is the default and --rna-
quantification-gc-bias does not have to be enabled. In addition, the library type is automatically
detected and --rna-quantification-library-type does not have to be set.
NOTE
Library-type auto-detection only works for paired-end data. If you have single-end data, you
need to specify the library type by setting the --rna-quantification-library-type option.
RNAFusion
To run gene fusion detection add the following option:
--enable-rna-gene-fusion true
You do not need to specify the library because fusion does not use it.
Epigenome Map and Align Examples
Prior to performing an epigenome (methylation) map and align run with bisulfite sequencing data, you
must first create methylation-specific reference hash tables, as follows:
mkdir -p /staging/human/reference/hg19_epigenome
dragen --build-hash-table true \
--ht-reference /staging/human/reference/hg19/hg19.fa \
--ht-max-seed-freq 64 --ht-seed-len 27 --ht-methylated true \
--output-directory /staging/human/reference/hg19_epigenome \
--ht-alt-liftover /opt/edico/liftover/hg19_alt_liftover.sam
The above dragen command produces two hash table directories under
/staging/human/reference/hg19_epigenome: GA_converted and CT_converted. The CT_
converted hash table is produced by converting each C base to T in the reference sequences.
Similarly, the GA_converted hash table is produced from the G->A base-converted reference
sequences. The base-converted references have less complexity, and to compensate, the hash table
seed length argument (--ht-seed-len) is typically increased to 27 for mammalian genomes (default
seed length is 21).
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
30
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The --ht-alt-aware-validate false option can be used in place of --ht-alt-liftover. However, the dragen
map quality will be significantly affected due to the presence of alternate contigs in the hg19.fa
reference.
Epigenome Map/Align, Directional-protocol, Single-Ended FASTQ Input, BAM
Output
The directional (Lister) protocol produces reads from two of the four possible bisulfite sequencing
strands. Therefore, when the --methylation-protocol=directional option is used, DRAGEN aligns each
read or read pair twice with different constraints corresponding to the two possible strands. The
following DRAGEN command produces two separate BAM files:
mkdir –p /staging/epigenome/directional
dragen -f –r /staging/human/reference/hg19_epigenome \
-1 /staging/epigenome/reads/sample_1_R1.fastq.gz \
-2 /staging/epigenome/reads/sample_10_R2.fastq.gz \
--RGID Illumina_RGID \
--RGSM sample_10 \
--RGPL illumina \
--output-directory /staging/epigenome/directional \
--output-file-prefix sample_10 \
--methylation-protocol=directional \
--enable-sort false
Epigenome Map/Align, Nondirectional-protocol, Paired-Ended FASTQ Input, BAM
Output
The nondirectional protocol produces reads from all four possible bisulfite sequencing strands.
Therefore, when the --methylation-protocol=non-directional argument is used, DRAGEN aligns each
read four times and produces four separate BAM files.
mkdir –p /staging/epigenome/non-directional
dragen -f –r /staging/human/reference/hg19_epigenome \
-1 /staging/epigenome/reads/sample_10_R1.fastq.gz \
-2 /staging/epigenome/reads/sample_10_R2.fastq.gz \
--RGID Illumina_RGID \
--RGSM sample_10 \
--RGPL illumina \
--output-directory /staging/epigenome/non-directional \
--output-file-prefix sample_10 \
--methylation-protocol non-directional \
--enable-sort false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
31
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Variant Calling Only Examples
The variant calling only examples show how you can pass an existing aligned BAM or CRAM file directly
to the DRAGEN Variant Caller. By default, the BAM/CRAM file is passed through the sorting stage prior
to variant calling.
To duplicate mark your BAM file before running the DRAGEN Variant Caller, you need to use a separate
tool. The DRAGEN Duplicate Marker depends on information provided by the Mapper/Aligner that does
not exist in BAM files. To take advantage of the DRAGEN Duplicate Marker, use DRAGEN in end-to-end
mode.
The BAM/CRAM files that are used as input to these example commands are not included in the
example data set. They are generated by example commands in Alignment Only Examples on page 26.
Unsorted BAM Input, VCF Output (Default)
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b /staging/examples/unsorted_SRA056922_30x_e10_50M.bam \
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix unsorted_output_SRA056922_30x_e10_50M \
--enable-map-align false
• Or, run /opt/edico/examples/unsorted_bam_in_vcf_out.sh.
Sorted BAM Input, VCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b /staging/examples/SRA056922_30x_e10_50M.bam \
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix sorted_output_SRA056922_30x_e10_50M \
--enable-map-align false
--enable-sort false
• Or, run /opt/edico/examples/sorted_bam_in_vcf_out.sh.
Sorted CRAM Input, VCF Output
• Enter the following input:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
32
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix sorted_output_SRA056922_30x_e10_50M \
--enable-sort false \
--enable-map-align false \
--cram-input /staging/examples/SRA056922_30x_e10_50M.cram
• Or, run /opt/edico/examples/sorted_cram_in_vcf_out.sh.
Somatic Small Variant Caller Examples
If you are using the Tumor-Normal BAM input option, and your BAMread groups have a shared RGID,
DRAGENcannot determine which read group the reads belong to. Ideally, you should have different
RGIDs for each read group, but you can work around the problem by setting the --prepend-filename-
to-rgid to true.
Paired-End FASTQ Input
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--tumor-fastq1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_
1.fastq.gz \
--tumor-fastq2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_
2.fastq.gz \
--enable-variant-caller true \
--RGID-tumor DRAGEN_RGID \
--RGSM-tumor DRAGEN_RGSM \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M
Sorted BAM Input
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--tumor-bam-input /staging/examples/SRA056922_30x_e10_50M.bam \
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix sorted_output_SRA056922_30x_e10_50M \
--enable-map-align false \
--prepend-filename-to-rgid true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
33
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
gVCF and Genotyping Examples
Paired-End FASTQ Input, gVCF Output
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--enable-variant-caller true \
--vc-emit-ref-confidence GVCF \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_e10_50M \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M
• Or, run /opt/edico/examples/paired_fastq_in_gVCF_out.sh.
Join Calling with gVCF Input
• Enter the following input:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--enable-joint-genotyping true \
--output-directory /staging/examples/ \
--output-file-prefix Joint_SRA056922_30x_e10_50M \
--variant /staging/examples/SRA056922_30x_e10_50M.gvcf.gz
• Or, run /opt/edico/examples/single_gVCF_in_jointVCF_out.sh.
Pedigree-Based Joint Genotyping with Three gVCF Files and a Pedigree File Input,
Joint-Genotyped VCF Output
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--enable-joint-genotyping true \
--output-directory /staging/examples/ \
--output-file-prefix Joint_SRA056922_30x_e10_50M \
--variant /staging/examples/mother.gvcf.gz \
--variant /staging/examples/father.gvcf.gz \
--variant /staging/examples/child.gvcf.gz \
--pedigree-file <PEGIGREE_FILE>
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
34
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

One Step Population-Based Joint Genotyping with gVCF Input, Joint-Genotyped
Multisample Output
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--enable-joint-genotyping true \
--output-directory /staging/examples/ \
--output-file-prefix Joint_SRA056922_30x_e10_50M \
--variant /staging/examples/SRA056922_30x_e10_50M.gvcf.gz
Two Step Population-Based Joint Genotyping with gVCF List Input, Joint-
Genotyped Multisample VCF Output
The first step generates a multisample VCF as output using a gVCF list as input and the following
command line option.
dragen -f \
--enable-gvcf-genotyper true \
--enable-map-align false \
--variant-list ${GVCF_LIST} \
--ht-reference ${FASTA_REF} \
--intermediate-results-dir ${TEMP_DIR} \
--output-directory ${OUTPUT_DIR} \
--output-file-prefix ${COHORT_NAME}
The second step generates a joint-genotyped multisample VCF using the multisample VCF produced
from step one as input and the following command line option. To specify the multisample VCF, use --
variant.
dragen -f \
--enable-joint-genotyping true \
--variant ${MULTISAMPLE_VCF} \
--ref-dir ${FASTA_REF} \
--output-directory ${OUTPUT_DIR}\
--output-file-prefix ${COHORT_NAME}.joint_genotyped
VCF to
generate
Population joint-
called
multisample
gVCF
Family joint-called
multisample gVCF
Population
joint-called
multisample
VCF
Family joint-
called
multisample
VCF
Table 2 Joint-calling modes, with associated input files and command line options.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
35
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Input file Multisample
combined gVCF
file
Multisample combined
gVCF file
Multi-sample
combined
gVCF file or X
individual gVCF
files
Multi-sample
combined
gVCF file or X
individual gVCF
files
Use
pedigree
file
No Yes No Yes
Command
line option
--enable-joint-
genotyping true -
-enable-multi-
sample-
gvcf=TRUE
--enable-joint-
genotyping true --
enable-multi-sample-
gvcf=TRUE --pedigree-
file file.ped
--enable-joint-
genotyping
true
--enable-joint-
genotyping
true --
pedigree-file
file.ped
Coverage Metrics Report Example
By default, DRAGEN reports read coverage over the whole genome, and if available also for the target
bed. You can specify up to three additional regions over which coverages are reported. In the following
example, DRAGEN generates two coverage reports, cov_report and full_res, for the additional
coverage region 1.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--RGID Illumina_RGID \
--RGSM SRA056922_30x_shuffle16k \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M \
--qc-coverage-region-1 /staging/examples/reads/vc_smoke.callable.bed \
--qc-coverage-reports-1 cov_report full_res
The full_res report corresponds to the Bedtools coverage option, and includes a per base resolution
read depth. The cov_report includes read depth summaries per region including mean, median, min
and max depths.
CNV Examples
These examples show how to use DRAGEN CNV to process already mapped and aligned BAM files
using the two modes of normalization supported by the DRAGEN CNV pipeline: Self Normalization and
Panel of Normals.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
36
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Self Normalization requires that the DRAGEN hash table be generated with the enable-cnv=true
option. It is recommended that you always generate a CNV compatible hash table if you frequently run
CNV.
The enable-map-align option is set to true by default in the configuration file. Set it to false if you do not
need to map and align the input BAM.
The --intermediate-results-dir option should be set to a local directory (eg, /staging/intermediate
or /local ssd). Otherwise, long processing time may occur during the CNV step.
Running with Self Normalization
Self normalization is the preferred method for single sample WGS processing. The BAM goes through
the entire CNV pipeline with a single command line.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b /staging/examples/SRA056922_30x_e10_50M.bam \
--intermediate-results-dir /staging/intermediate \
--output-directory /staging/examples/ \
--output-file-prefix dragen_cnv1 \
--enable-map-align false \
--enable-cnv true \
--cnv-enable-self-normalization true \
Running with a Panel of Normals
The Panel of Normals approach requires pregenerating the target.counts file for each sample to be
used, and then executing one final command to perform the normalization and copy number variant
calling.
To calculate target counts with BAM Input, this example command extracts the signals, including read
counts, from the alignments of the BAM file. It generates a *.target.counts file to be used in the
normalization step. Target counts should be calculated for each input BAM file, including the case
sample under analysis and the normals samples.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b /staging/examples/SRA056922_30x_e10_50M.bam \
--intermediate-results-dir /staging/intermediate \
--output-directory /staging/examples/ \
--output-file-prefix dragen_cnv1 \
--enable-map-align false \
--enable-cnv true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
37
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The following command performs the normalization and generates the CNV calls. The normals
samples should be listed in a text file (normal.txt in this example) that provides the path to the
*.target.counts files of the normal samples. The case sample *.target.counts file is specified with the --
cnv-input option. In this example, gcbias correction of the input is disabled.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--intermediate-results-dir /staging/intermediate \
--output-directory /staging/examples/ \
--output-file-prefix dragen_cnv2 \
--enable-cnv true \
--cnv-input /staging/examples/dragen_cnv1.target.counts \
--cnv-normals-list normal.txt \
--cnv-enable-gcbias-correction false
FASTQ Processing
This example runs the DRAGEN CNV caller in Self Normalization mode directly from FASTQ samples. It
first maps and aligns the FASTQ and continues directly to CNV calling. This step can be combined with
variant calling.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_1.fastq.gz \
-2 /staging/examples/reads/SRA056922_30x_shuffle16k_e10_50M_2.fastq.gz \
--RGID Illumina_ID \
--RGSM SRA056922_30x_shuffle16k \
--intermediate-results-dir /staging/intermediate \
--output-directory /staging/examples/ \
--output-file-prefix dragen_cnv \
--enable-map-align true \
--enable-cnv true \
--cnv-enable-self-normalization true
Running De Novo CNV Calling
De novo calling requires previously generated normalized signal files (*.tn.tsv) from the single sample
analysis. If a pedigree file is supplied, then a de novo state and a de novo quality score will be
annotated for the proband sample’s records.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--cnv-input father.tn.tsv \
--cnv-input mother.tn.tsv \
--cnv-input child.tn.tsv \
--intermediate-results-dir /staging/intermediate \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
38
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-directory /staging/examples/ \
--output-file-prefix trio_cnv \
--pedigree-file trio.ped \
--enable-cnv true
Running Somatic CNV Calling
Somatic CNV calling requires a tumor and matched normal sample. The matched normal sample must
first go through the germline small variant caller to produce a *.hard-filtered.vcf.gz. If known, it is
recommended that you specify the sample sex.
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--tumor-bam-input tumor.bam \
--intermediate-results-dir /staging/intermediate \
--output-directory /staging/examples/ \
--output-file-prefix somatic_cnv \
--enable-map-align false \
--enable-cnv true \
--cnv-normal-b-allele-vcf normal.hard-filtered.vcf.gz \
--sample-sex female
Structural Variant Calling Examples
Structural Variant calling can run in the following modes:
• Standalone—Runs from mapped BAM/CRAM input files. Requires the --enable-map-align=false
and --enable-sv=true options.
• Integrated—Automatically runs on the output of the DRAGEN mapper/aligner. Requires the
--enable-map-align=true, --enable-sv=true, and --enable-map-align-output=true options.
Structural Variant calling can be enabled along with any other caller as well.
Integrated Execution Example
dragen -f \
--ref-dir <HASH_TABLE_DIR> \
--enable-map-align true \
--enable-map-align-output true \
--enable-sv true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX> \
-1 <FASTQ1> -2 <FASTQ2> \
--RGID <RGID> \
--RGSM <RGSM>
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
39
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Standalone Joint Diploid Calling Example
dragen -f \
--ref-dir <HASH_TABLE_DIR> \
--enable-map-align false \
--enable-sv true \
--bam-input <BAM1> \
--bam-input <BAM2> \
--bam-input <BAM3> \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX>
Standalone De Novo Quality Scoring Example
dragen -f \
--variant <TRIO_VCF_FILE> \
--pedigree-file <PED_FILE> \
--enable-map-align false \
--sv-denovo-scoring true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX>
BCL to FASTQ Conversion with Minimal Settings
This example shows how to use DRAGEN to process Illumina BCL format files.
The BCL directory used in the example is not included in the example data package. Replace
/mnt/san/131022_hsxten008_0123_FC543 with your own BCL directory.
• Enter the following input:
dragen --bcl-conversion-only=true \
--bcl-input-directory /mnt/san/131022_hsxten008_0123_FC543 \
-- output-directory /staging/examples/
• Or run /opt/edico/examples/bcl_in_fastq_out.sh.
S3 and HTTP Streaming Input Examples
DRAGEN can process input files directly from an S3 bucket, or using HTTP presigned URLs, which is
known as input streaming. The input files do not need to be downloaded to a local disk prior to being
processed. Instead, the files are streamed over the network directly into the DRAGEN processor.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
40
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Streaming is supported for compressed FASTQ (*.fastq.gz) files. A future version of DRAGEN will also
support streaming from BAM (*.bam) files. Input streaming can be used in all the configurations that
use single-end FASTQs, paired-end FASTQs, and FASTQ lists. The following examples show some of
the ways to use input streaming.
Streaming FASTQ Input using S3
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 s3://s3-bucket-name/path/to/object_1.fastq.gz \
-2 s3://s3-bucket-name/path/to/object_2.fastq.gz \
--RGID object_ID \
--RGSM sample_name \
--output-directory /staging/examples/ \
--output-file-prefix streaming
Streaming FASTQ Input using HTTP
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 https://bucket-name.amazonaws.com/path/to/object_1.fastq.gz?querystring
\
-2 https://bucket-name.amazonaws.com/path/to/object_2.fastq.gz$querystring
\
--RGID object_ID \
-RGSM sample_name \
--output-directory /staging/examples/ \
--output-file-prefix streaming
You need to have permission to access the remote files. If you have access to the file, then DRAGEN is
capable of streaming the remote file. The S3 object requires AWS authentication and credentials. The
authentication should already be set up on the instance you are running, for example, via IAM policies.
An HTTP URL most likely has a query string attached to it, which provides the authentication
credentials or necessary tokens to grant permission. The security method may be present in other
parts of the URL, for example:
https://stagingdl.dnanex.us/security/string/sample_1.fastq.gz
Multicaller Workflows
DRAGEN supports running multiple tools in a single workflow.
The enable-component flag controls which components are enabled or disabled. DRAGEN constructs a
workflow using the enabled components and automatically resolves any component inconsistencies.
When possible, DRAGEN runs components in parallel.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
41
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Each component has a set of options that configures input settings, internal algorithm parameters, or
output files and filtering criteria. Refer to the individual component sections for more details.
Some options, such as output-directory and sample-sex, are shared amongst callers.
Each variant caller produces its own set of VCFs and metric output files.
Example Component Commands
enable-map-align
enable-sort
enable-duplicate-marking
enable-variant-caller
enable-cnv
enable-sv
Input Formats
DRAGEN accepts the following common standard NGS input formats:
• FASTQ (fastq-file1 and fastq-file2)
• FASTQ List (fastq-list)
• BAM (bam-input)
• CRAM (cram-input)
Somatic workflows can use tumor equivalent input files (eg, fastq-file1 and fastq-file2).
When running from unaligned reads, the reads first go through the map/align component to produce
alignments which continue downstream to the variant callers. When running from prealigned reads,
DRAGEN supports re-aligning with the map/align component or using the existing alignments from the
source input.
Multicaller Command Line Example
The following example demonstrates best practices for combining command line options from single
caller scenarios to create a multicaller workflow. The example consists of the following steps:
• Configure the INPUT options.
• Configure the OUTPUT options.
• Configure MAP/ALIGN depending on if realignment is desired or not.
• Configure the VARIANT CALLERs based on the application.
• Build up the necessary options for each component separately, to enable reuse in the final
command line.
INPUT_OPTIONS="
--ref-dir $DRAGEN_HASH_TABLE \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
42
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--fastq-file1 $FASTQ1 \
--fastq-file2 $FASTQ2 \
--RGSM $RGSM \
--RGID $RGID \
"
OUTPUT_OPTIONS="
--output-directory $OUTPUT \
--output-file-prefix $PREFIX \
"
MA_OPTIONS="
--enable-map-align true \
... <any other optional settings> \
"
CNV_OPTIONS="
--enable-cnv true \
... <any other optional settings> \
"
SNV_OPTIONS="
--enable-variant-caller true \
... <any other optional settings> \
"
SV_OPTIONS="
--enable-sv true \
... <any other optional settings> \
"
CMD="
dragen \
$INPUT_OPTIONS \
$OUTPUT_OPTIONS \
$MA_OPTIONS \
$CNV_OPTIONS \
$SNV_OPTIONS \
$SV_OPTIONS \
"
Germline
The following table summarizes the support for some input formats and variant callers. Some
supported features and callers are not listed in the table.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
43
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

GERMLINE FASTQ with Map/Align BAM/CRAM BAM/CRAM with Map/Align
CNV + SNV Supported Supported Supported
CNV + SV Supported Supported Supported
SNV + SV Supported Supported Supported
CNV + SNV + SV Supported Supported Supported
Somatic
Somatic workflows specify both tumor and normal inputs. The need for potentially two input files
(tumor and matched normal) as well as the need for a matched normal SNV VCF for the Somatic CNV
caller means extra care has to be taken. For this reason, the recommended tumor/normal workflow
first starts with running the matched normal through the Germline Workflow.
1. Run matched normal through Germline workflow (CNV + SNV + SV + ...). The workflow generates
the matched normal SNV VCF.
2. Run tumor and matched normal through Somatic workflow (CNV + SNV + SV + ...)
INPUT_OPTIONS="
--ref-dir $DRAGEN_HASH_TABLE \
--tumor-bam-input $TUMOR_BAM \
--bam-input $NORMAL_BAM \
"
OUTPUT_OPTIONS="
--output-directory $OUTPUT \
--output-file-prefix $PREFIX \
"
MA_OPTIONS="
--enable-map-align false \
... <any other optional settings> \
"
CNV_OPTIONS="
--enable-cnv true \
--cnv-normal-b-allele-vcf $SNV_VCF \
... <any other optional settings> \
"
SNV_OPTIONS="
--enable-variant-caller true \
... <any other optional settings> \
"
SV_OPTIONS="
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
44
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

--enable-sv true \
... <any other optional settings> \
"
CMD="
dragen \
$INPUT_OPTIONS \
$OUTPUT_OPTIONS \
$MA_OPTIONS \
$CNV_OPTIONS \
$SNV_OPTIONS \
$SV_OPTIONS \
"
The following table lists the various combinations that are supported under the tumor/normal mode of
operation.
Tumor normal FASTQ with Map/Align BAM/CRAM BAM/CRAM with Map/Align
CNV + SNV Supported Supported Supported
CNV + SV Supported Supported Supported
SNV + SV Supported Supported Supported
CNV + SNV + SV Supported Supported Supported
To run in tumor only mode, remove the matched normal input from the INPUT options and configure
each individual caller to run in tumor only mode. The following table lists the combinations that are
supported under the tumor only mode of operation.
Tumor normal FASTQ with Map/Align BAM/CRAM BAM/CRAM with Map/Align
CNV + SNV Supported Supported Supported
CNV + SV Supported Supported Supported
SNV + SV Supported Supported Supported
CNV + SNV + SV Supported Supported Supported
WES analysis is supported if the mode is supported in singe caller mode and there is no input
configuration conflict.
Troubleshooting
The DRAGEN software automatically resets the board if any problems are encountered. In the rare
case that reset does not occur automatically, you can use the following command to reset:
/opt/edico/bin/dragen_reset
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
45
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
If resetting does not resolve the issue, contact Illumina Technical Support. Run the following command
to collect diagnostic and configuration information for Technical Support.
sudo sosreport --batch --tmp-dir /staging/tmp
This tool takes several minutes to run and reports the location where it has saved the diagnostic
information in /staging/tmp. Please include the report when you submit a support ticket for Illumina
Technical Support.
For more information, refer to the DRAGEN Bio-IT Platform User Guide(1000000070494) on the
Illumina Support Site.
Additional Resources and Support
For additional information, resources, system updates, and support, please visit the DRAGEN support
page on the Illumina website.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
46
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
DRAGEN Host Software
You use the DRAGENhost software program dragen to build and load reference genomes, and then to
analyze sequencing data by decompressing the data, mapping, aligning, sorting, duplicate marking
with optional removal, and variant calling.
Invoke the software using the dragen command. The command-line options are described in the
following sections.
Command-line options can also be set in a configuration file.For more information on configuration
files, see Configuration Files on page 61. If an option is set in the configuration file and is also specified
on the command-line, the command-line option overrides the configuration file.
Command-line Options
For a complete list of command line options, see Command Line Options on page 1.
DRAGEN Command-Line Options
Perform the following command-line options using the dragen:
• Build Reference/Hash Table
dragen --build-hash-table true --ht-reference <REF_FASTA> \
--output-directory <REF_DIRECTORY> [options]
• Run Map/Align and Variant Caller (*.fastq to *.vcf)
dragen -r <REF_DIRECTORY> --output-directory <OUT_DIRECTORY> \
--output-file-prefix <FILE_PREFIX> [options] -1 <FASTQ1> \
[-2 <FASTQ2>] –-RGID <RG0> --RGSM <SM0> --enable-variant-caller true
• Run Map/Align (*.fastq to *.bam)
dragen -r <REF_DIRECTORY> --output-directory <OUT_DIRECTORY> \
--output-file-prefix <FILE_PREFIX> [options] \
-1 <FASTQ1> [-2 <FASTQ2>] \
--RGID <RG0> --RGSM
• Run Variant Caller (*.bam to *.vcf)
dragen -r <REF_DIRECTORY> --output-directory <OUT_DIRECTORY> \
--output-file-prefix <FILE_PREFIX> [options] -b <BAM> \
--enable-variant-caller true
• Run BCL Converter (BCL to *.fastq)
dragen --bcl-conversion-only true --bcl-input-directory <BCL_DIRECTORY> \
--output-directory <OUT_DIRECTORY>
• Run RNA Map/Align (*.fastq to *.bam)
dragen -r <REF_DIRECTORY> --output-directory <OUT_DIRECTORY> \
--output-file-prefix <FILE_PREFIX> [options] -1 <FASTQ1> \
[-2 <FASTQ2>] --enable-rna true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
47
Operating Modes
DRAGEN has two primary modes of operation, as follows:
• Mapper/aligner
• Variant caller
DRAGEN is capable of performing each mode independently or as an end-to-end solution.
DRAGENalso allows you to enable and disable decompression, sorting, duplicate marking, and
compression along the DRAGENpipeline.
Full Pipeline Mode
To execute full pipeline mode, set --enable-variant-caller to true and provide input as unmapped reads
in *.fastq, *.bam, or *.cram formats.
DRAGENperforms decompression, mapping, aligning, sorting, and optional duplicate marking and
feeds directly into the variant caller to produce a VCF file. In this mode, DRAGENuses parallel stages
throughout the pipeline to drastically reduce the overall run time.
Map/Align Mode
Map/align mode is enabled by default. Input is unmapped reads in *.fastq, *.bam, or *.cram format.
DRAGENproduces an aligned and sorted BAM or CRAM file. To mark duplicate reads at the same time,
set --enable-duplicate-marking to true.
Variant Caller Mode
To execute variant caller mode, set the --enable-variant-caller option to true.
Input is a mapped and aligned BAM file. DRAGENproduces a VCF file. If the BAM file is already sorted,
you can skip sorting by setting --enable-sort to false. BAM files cannot be duplicate marked in the
DRAGENpipeline prior to variant calling if they have not already been marked. Use the end-to-end
mode of operation to take advantage of the mark-duplicates feature.
RNA-Seq Data
To enable processing of RNA-Seq–based data, set --enable-rna to true.
DRAGEN uses the RNA spliced aligner during the mapper/aligner stage. DRAGEN dynamically switches
between the required modes of operation.
Bisulfite MethylSeq Data
To enable processing of Bisulfite MethylSeq data, set the --enable-methylation-calling option to true.
DRAGEN automates the processing of data for Lister and Cokus (aka directional and nondirectional)
protocols, generating a single BAM with bismark-compatible tags.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
48
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Alternatively, you can run DRAGENin a mode that produces a separate BAM file for each combination
of the C->T and G->A converted reads and references.To enable this mode of processing, you need
to build a set of reference hash tables with --ht-methylated enabled, and run dragen with the
appropriate --methylation-protocol setting.
Reference Genome Options
Before you can use the DRAGENsystem for aligning reads, you must load a reference genome and its
associated hash tables onto the PCIe card.For information on preprocessing a reference genome’s
FASTA files into the native DRAGEN binary reference and hash table formats, see Prepare a Reference
Genome on page 8. You must also specify the directory containing the preprocessed binary reference
and hash tables with the -r [or --ref-dir] option. This argument is always required.
Use the following command to You can load the reference genome and hash tables to DRAGEN card
memory separately from processing reads/, as follows.
dragen -r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149
You can Uuse the -l (--force-load-reference) option to force the reference genome to load even if it is
already loaded, as follows.
dragen -l -r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149
The time needed to load the reference genome depends on the size of the reference, but for typical
recommended settings, it takes approximately 30–60 seconds.
Output Options
The following commandline options for output are mandatory:
• --output-directory <out_dir>—Specifies the output directory for generated files.
• --output-file-prefix <out_prefix>—Specifies the output file prefix. DRAGENappends the
appropriate file extension onto this prefix for each generated file.
• -r [ --ref-dir ] specifies the reference hash table.
For brevity, the following examples do not include these mandatory options.
For mapping and aligning, the output is sorted and compressed into BAM format by default before
saving to disk. You can control the output format from the map/align stage with the --output-format
<SAM|BAM|CRAM> option. If the output file exists, DRAGENissues a warning and exits. To force
overwrite if the output file already exists, use the -f [ --force ] option.
For example, the following commands output to a compressed BAM file and forces overwrite:
dragen ... -f
dragen ... -f --output-format bam
To generate a BAI-format BAM index file (.bai file extension), set --enable-bam-indexing to true.
The following example outputs to a SAM file and forces overwrite:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
49
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

dragen ... -f --output-format sam
The following example outputs to a CRAM file and forces overwrite:
dragen ... -f --output-format cram
DRAGENcan generate mismatch difference (MD) tags, as described in the BAM standard.The feature
is turned off by default because there is a small performance cost to generate these strings.To
generate MDtags, set --generate-md-tags to true.
To generate ZS:Z alignment status tags, set --generate-zs-tags to true.These tags are only
generated in the primary alignment and when a read has suboptimal alignments qualifying for
secondary output (even if none were output because --Aligner.sec-aligns was set to 0).
The following are valid tag values:
• ZS:Z:R—Multiple alignments with similar score were found.
• ZS:Z:NM—No alignment was found.
• ZS:Z:QL—An alignment was found, but it was below the quality threshold.
To generate SA:Z tags, set --generate-sa-tags to true (the default). These tags provide alignment
information (position, cigar, orientation) of groups of supplementary alignments, which are useful in
structural variant calling.
Preservation or Stripping of BQSR Tags
The Picard Base Quality Score Recalibration (BQSR) tool produces output BAM files that include tags BI
and BD.BQSR calculates these tags relative to the exact sequence for a read.If a BAM file with BIand
BD tags is used as input to mapper/aligner with hard clipping enabled, the BI and/or BD tags can
become invalid.
The recommendation is to strip these tags when using BAM files as input.To remove the BI and BD
tags, set the --preserve-bqsr-tags option to false.If you preserve the tags, DRAGENwarns you to
disable hard clipping.
Read Group Options
DRAGEN assumes that all the reads in a given FASTQ belong to the same read group. The DRAGEN
system creates a single @RG read group descriptor in the header of the output BAM file, with the ability
to specify the following standard BAM attributes:
Attribute Argument Description
ID --RGID Read group identifier.If you include any of the read
group parameters, RGID is required. It is the value
written into each output BAM record.
LB --RGLB Library.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
50
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Attribute Argument Description
PL --RGPL Platform/technology used to produce the reads.The
BAM standard allows for values CAPILLARY, LS454,
ILLUMINA, SOLID, HELICOS, IONTORRENT and
PACBIO.
PU --RGPU Platform unit, eg, flowcell-barcode.lane.
SM --RGSM Sample.
CN --RGCN Name of the sequencing center that produced the
read.
DS --RGDS Description.
DT --RGDT Date the run was produced.
PI --RGPI Predicted mean insert size.
If any of these arguments are present, the DRAGEN software adds an RG tag to all the output records
to indicate that they are members of a read group.The following example shows a command line that
includes read group parameters:
dragen --RGID 1 --RGCN Broad --RGLB Solexa-135852 \
--RGPL Illumina --RGPU 1 --RGSM NA12878 \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 SRA056922.fastq --output-directory /staging/tmp/ \
--output-file-prefix rg_example
When using the --fastq-list option to input multiple read groups, BAM tags (and others) are specified
for each read group by adding columns to the fastq_list.csv file. Each column heading consists of
four capital letters and each begins with 'RG'. For each column, each read group’s values for that
column are propagated to the output BAM file in an identically named tag.
License Options
To suppress the license status message at the end of the run, use the --lic-no-print option. The
following shows an example of the license status message:
LICENSE_MSG| =====================================================
LICENSE_MSG| License report
LICENSE_MSG| Genome status [ACxxxxxxxxxxx] : used 1263.9 Gbases since
2018-Feb-15 (1263886160894 bases, unlimited)
LICENSE_MSG| Genome bases [ACxxxxxxxxxxx] : 202000000
LICENSE_MSG| Genome bases [total] : 202000000
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
51
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Input Options
The DRAGEN system is capable of processing reads in FASTQ format or BAM/CRAM format. If your
input FASTQ files end in .gz, DRAGEN automatically decompresses the files using hardware-
accelerated decompression.
Input File Types
Use the following command-line options for any input files you would like to use.
FASTQ Input Files
FASTQ input files can be single-ended or paired-end. Use the following examples to input FASTQ files.
• Single-ended in one FASTQ file (-1 option)
dragen -r <REF_DIR> -1 <fastq> --output-directory <OUT_DIR> \
--output-file-prefix <OUTPUT_PREFIX> --RGID <RGID> --RGSM <RGSM>
• Paired-end in two matched FASTQ files(-1 and -2 options)
dragen -r <REF_DIR> -1 <fastq1> -2 <fastq2> \
--output-directory <OUT_DIR> --output-file-prefix <OUT_PREFIX> \
--RGID <RGID> --RGSM <RGSM>
• Paired-end in a single interleaved FASTQ file(--interleaved (-i) option)
dragen -r <REF_DIR> -1 <INTERLEAVED_FASTQ> -i \
--RGID <RGID> --RGSM <RGSM>
If using, bcl2fastq or the DRAGEN BCL command use the following common file naming convention:
<SampleID>_S<#>_<Lane>_<Read>_<segment#>.fastq.gz
The file naming convention is different for HiSeq X and NextSeq Sequencing Systems.
Older versions of bcl2fastq and DRAGENcould segment FASTQ samples into multiple files to limit file
size or to decrease the time to generate them.
For Example:
RDRS182520_S1_L001_R1_001.fastq.gz
RDRS182520_S1_L001_R1_002.fastq.gz
...
RDRS182520_S1_L001_R1_008.fastq.gz
These files do not need to be concatenated to be processed together by DRAGEN. To map/align any
sample, provide the first file in the series (-1 <FileName>_001.fastq). DRAGENreads all segment files in
the sample consecutively for both of the FASTQ file sequences specified using the -1 and -2 options for
paired-end input and for compressed fastq.gz files. To turn the behavior off, set --enable-auto-
multifile to false on the command line.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
52
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
DRAGENcan also optionally read multiple files by the sample name given in the file name, which can be
used to combine samples that have been distributed across multiple BCL lanes or flow cells. To enable
this feature, set the --combine-samples-by-name option to true.
If the FASTQ files specified on the command-line use the Casava 1.8 file naming convention shown
above and additional files in the same directory share that sample name, those files and all their
segments are processed automatically. Note that sample name, read number, and file extension must
match. Index barcode and lane number can differ.
To avoid impacting system performance, input files must be located on a fast file system.
Multiple FASTQ Input Files
To provide multiple FASTQ input files, it is recommended to use the --fastq-list <csv file name> option
to specify the name of a CSV file containing the list of FASTQ files, instead of using the --combine-
samples-by-name option. For example:
dragen -r <ref_dir> --fastq-list <CSV_FILE> \
--fastq-list-sample-id <Sample_ID> \
--output-directory <OUT_DIR> --output-file-prefix <OUT_PREFIX>
Using a CSVfile allows you to name the FASTQ input files, input from multiple subdirectories, and add
BAM tags specified explicitly for each read group. DRAGENautomatically generates a CSV file of the
correct format during BCL conversion to FASTQ. The CSVfile is named fastq_list.csv and contains
an entry for each FASTQ file or paired-end file pair produced during the run.
FASTQ CSV File Format
The first line of the CSV file specifies the title of each column, and is followed by one or more data lines.
All lines in the CSV file must contain the same number of comma-separated values and should not
contain white space or other extraneous characters.
Column titles are case-sensitive. The following column titles are required:
• RGID—Read Group
• RGSM—Sample ID
• RGLB—Library
• Lane—Flow cell lane
• Read1File—Full path to a valid FASTQ input file
• Read2File—Full path to a valid FASTQ input file. Required for paired-end input. If not using paired-
end input, leave empty.
Each FASTQ file can only be referenced once in the CSV list.. All values in the Read2File column must
be reference valid files or must all be empty.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
53
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

When generating a BAM file using fastq-list input, one read group is generated per unique RGID value.
The BAM header contains RG tags for the following read groups:
• ID (from RGID)
• SM (from RGSM)
• LB (from RGLB)
Additional tags can be specified for each read group by adding a column title. The column title must be
only four upper-case characters and begin with RG. For example, to add a PU (platform unit) tag, add a
column named RGPU and specify the value for each read group in this column. All column titles must be
unique.
A fastq-list file can contain files for more than one sample. If a fastq-list file contains only one unique
RGSM entry, then no additional options need to be specified and DRAGEN processes all files listed in
the fastq-list file. If there is more than one unique RGSM entry in a fastq-list file, one of the following
must also be specified in addition to --fastq-list <filename>.
• To process a specific sample from the CSVfile, use --fastq-list-sample-id <SampleID>. Only the
entries in the fastq-list file with an RGSMvalue matching the specified SampleID are processed.
• To process all samples together in the same run, regardless of the RGSMvalue, set --fastq-list-all-
samples to true.
For a single run, only one BAM and VCF output file are produced because all input read groups
are expected to belong to the same sample. To process multiple samples from one BCL
conversion run, run the DRAGEN secondary analysis multiple times using different values for the
--fastq-list-sample-id option.
There is no option to specify groupings or subsets of RGSM values for more complex filtering, but the
fastq-list file can be modified to achieve the same effect.
The following is an example FASTQ list CSV file with the required columns:
RGID,RGSM,RGLB,Lane,Read1File,Read2File
CACACTGA.1,RDSR181520,UnknownLibrary,1,/staging/RDSR181520_S1_L001_R1_
001.fastq, /staging/RDSR181520_S1_L001_R2_001.fastq
AGAACGGA.1,RDSR181521,UnknownLibrary,1,/staging/RDSR181521_S2_L001_R1_
001.fastq, /staging/RDSR181521_S2_L001_R2_001.fastq
TAAGTGCC.1,RDSR181522,UnknownLibrary,1,/staging/RDSR181522_S3_L001_R1_
001.fastq, /staging/RDSR181522_S3_L001_R2_001.fastq
AGACTGAG.1,RDSR181523,UnknownLibrary,1,/staging/RDSR181523_S4_L001_R1_
001.fastq, /staging/RDSR181523_S4_L001_R2_001.fastq
If you use the --tumor-fastq-list option for somatic input, use the --tumor-fastq-list-sample-id
<SampleID> option to specify the sample ID for the corresponding FASTQ list, as shown in the
following example:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
54
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
dragen -r <ref_dir> --tumor-fastq-list <csv_file> \
--tumor-fastq-list-sample-id <Sample_ID> \
--output-directory <out_dir> \
--output-file-prefix <out_prefix> --fastq-list <csv_file_2> \
--fastq-list-sample-id <Sample_ID_2>
Tumor-Normal Pairs Input
If using fastq_lists or tumor_fastq_lists comprising of multiple samples (RGSMs) in somatic mode, you
can use a loop to iterate through the two lists to create tumor-normal pairs for testing. Create a *.txt
file with the RGSM of each normal sample to be tested (one per line), and then create a separate *.txt
file with the RGSM of the tumor samples to be tested. Make sure that the tumor sample RGSM is listed
in the same order as the corresponding normal samples and to include a blank line after the last
sample.
The following example script can then be used to perform testing in somatic mode. Each iteration takes
one entry from the tumor samples list and one entry from the normal samples list (from top to bottom)
to create a tumor-normal pair as input for the DRAGEN run.
#!/bin/bash
HT="/staging/HT/"
tumor_fastq_list="/staging/inputs/tumor_fastq_list.csv"
normal_fastq_list="/staging/inputs/normal_fastq_list.csv"
tumor_samples_list="/staging/inputs/tumor_samples_list.txt"
normal_samples_list="/staging/inputs/normal_samples_list.txt"
while read -u 3 -r tumor_RGSM && read -u 4 -r normal_RGSM; do
output_dir="/staging/results/${tumor_RGSM}_${normal_RGSM}"
mkdir -p ${output_dir}
dragen \
-r ${HT} \
--tumor-fastq-list ${tumor_fastq_list} \
--tumor-fastq-list-sample-id ${tumor_RGSM} \
--fastq-list ${normal_fastq_list} \
--fastq-list-sample-id ${normal_RGSM} \
--output-directory ${output_dir} \
--output-file-prefix ${tumor_RGSM}_${normal_RGSM}
done 3<${tumor_samples_list} 4<${normal_samples_list}
The following are examples of the FASTQ lists and samples lists used as input for the script.
Sample fastq_list.csv:
RGPL,RGID,RGSM,RGLB,Lane,Read1File,Read2File
DRAGEN_RGPL,DRAGEN_RGID_N1.1,normal-1,ILLUMINA,1,/staging/inputs/normal-1_
S1_L001_R1_001.fastq.gz,/staging/inputs/normal-1_S1_L001_R2_001.fastq.gz
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
55
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
DRAGEN_RGPL,DRAGEN_RGID_N1.2,normal-1,ILLUMINA,2,/staging/inputs/normal-1_
S1_L002_R1_001.fastq.gz,/staging/inputs/normal-1_S1_L002_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_N2.1,normal-2,ILLUMINA,1,/staging/inputs/normal-2_
S1_L001_R1_001.fastq.gz,/staging/inputs/normal-2_S1_L001_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_N2.2,normal-2,ILLUMINA,2,/staging/inputs/normal-2_
S1_L002_R1_001.fastq.gz,/staging/inputs/normal-2_S1_L002_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_N3.1,normal-3,ILLUMINA,1,/staging/inputs/normal-3_
S1_L001_R1_001.fastq.gz,/staging/inputs/normal-3_S1_L001_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_N3.2,normal-3,ILLUMINA,2,/staging/inputs/normal-3_
S1_L002_R1_001.fastq.gz,/staging/inputs/normal-3_S1_L002_R2_001.fastq.gz
Sample tumor_fastq_list.csv content:
RGPL,RGID,RGSM,RGLB,Lane,Read1File,Read2File
DRAGEN_RGPL,DRAGEN_RGID_T1.1,tumor-1,ILLUMINA,1,/staging/inputs/tumor-1_
S1_L001_R1_001.fastq.gz,/staging/inputs/tumor-1_S1_L001_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_T1.2,tumor-1,ILLUMINA,2,/staging/inputs/tumor-1_
S1_L002_R1_001.fastq.gz,/staging/inputs/tumor-1_S1_L002_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_T2.1,tumor-2,ILLUMINA,1,/staging/inputs/tumor-2_
S1_L001_R1_001.fastq.gz,/staging/inputs/tumor-2_S1_L001_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_T2.2,tumor-2,ILLUMINA,2,/staging/inputs/tumor-2_
S1_L002_R1_001.fastq.gz,/staging/inputs/tumor-2_S1_L002_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_T3.1,tumor-3,ILLUMINA,1,/staging/inputs/tumor-3_
S1_L001_R1_001.fastq.gz,/staging/inputs/tumor-3_S1_L001_R2_001.fastq.gz
DRAGEN_RGPL,DRAGEN_RGID_T3.2,tumor-3,ILLUMINA,2,/staging/inputs/tumor-3_
S1_L002_R1_001.fastq.gz,/staging/inputs/tumor-3_S1_L002_R2_001.fastq.gz
Sample normal_samples_list
normal-1
normal-2
normal-3
Sample tumor_samples_list content
tumor-1
tumor-2
tumor-3
BAM Input Files
To use BAM files as input to the mapper/aligner, set --enable-map-align to true. If you leave this option
set to false (the default), you can use the BAM file as input to the variant caller.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
56
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
When you specify a BAM file as input, DRAGEN ignores any alignment information contained in the
input file, and outputs new alignments for all reads.If the input file contains paired-end reads, it is
important to specify that the input data should be sorted so that pairs can be processed
together.Other pipelines require you to resort the input data set by read name. DRAGEN vastly
increases the speed of this operation by pairing the input reads, and sending on to the mapper/aligner
when pairs are identified. Use the --pair-by-name option to enable or disable this feature (the default
is true).
• Specify single-ended input in one BAM file with the (-b) and --pair-by-name=false options, as
follows:
dragen -r <ref_dir> -b <bam> --output-directory <out_dir> \
--output-file-prefix <out_prefix> --pair-by-name false
• Specify paired-end input in one BAM file with the (-b) and --pair-by-name=true options, as
follows:
dragen -r <ref_dir> -b <bam> --output-directory <out_dir> \
--output-file-prefix <out_prefix> --pair-by-name true
CRAM Input Files
You can use CRAM files as input to the DRAGEN mapper/aligner and variant caller. The DRAGEN
functionality available when using CRAM input is the same as when using BAM input.
The --cram-reference option is no longer needed. The CRAM compressor and decompressor uses the
DRAGEN reference.
Use the following options to provide a CRAM input to either mapper/aligner or variant caller:
• --cram-input—The name and path for the CRAM file.
• --cram-input—One usage example is paired-end input in a single CRAM file. In addition, set the --
pair-by-name option to true.
dragen -r <ref_dir> --cram-input <cram> --output-directory <out_dir> \
--output-file-prefix <out_prefix> --pair-by-name true
BCL Input Files
BCL is the output format of Illumina sequencing systems. Under limited circumstances, DRAGEN can
read directly from BCL for map-align operations, saving the time needed for conversion to FASTQ.
DRAGEN can read directly from BCL in the following circumstances:
• Only one lane is input as part of a run (specified on the command-line).
• The lane has only a single sample specified in the SampleSheet.csv file.
When converting BCLto FASTQ is required, DRAGEN provides a BCL to FASTQ converter (see DRAGEN
BCL Data Conversion on page 238).
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
57
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The following example command is for BCL input with only one lane of input:
dragen --bcl-input-dir <BCL_ROOT> --bcl-only-lane <num> -r <ref_dir> \
--output-directory <out_dir> --output-file-prefix <out_prefix>
For additional BCL conversion options, see DRAGEN BCL Data Conversion on page 238.
Handling of N bases
One of the techniques that DRAGEN uses to optimize handling sequences can lead to the overwriting
the base quality score assigned to N base calls.
When you use the --fastq-n-quality and --fastq-offset options, the base quality scores are overwritten
with a fixed base quality.The default values for these options are 2 and 33 respectively to match the
Illumina minimum quality of 35 (ASCII character ‘#’).
Read Names for Paired-End Reads
By a common convention, read names can include suffixes (such as /1 or /2), which indicate the end of
a pair the read represents. For BAM input using the --pair-by-name option, DRAGEN ignores these
suffixes to find matching pair names. By default, DRAGEN uses the forward slash character as the
delimiter for these suffixes and ignores the /1 and /2 when comparing names. By default, DRAGEN
strips these suffixes from the original read names.
DRAGEN provides the following options to control how suffixes are used:
• To change the delimiter character for suffixes, use the --pair-suffix-delimiter option. Valid values
for this option include forward-slash (/), dot (.), and colon (:).
• To preserve the entire name, including the suffixes, set --strip-input-qname-suffixes to false.
• To append a new set of suffixes to all read names, set --append-read-index-to-name to true,
where the delimiter is determined by the --pair-suffix-delimiter option.By default the delimiter is a
slash, so /1 and /2 are added to the names.
Gene Annotation Input Files
When processing RNA-Seq data, you can supply a gene annotations file by using the --annotation-file
option. Providing this file improves the accuracy of the mapping and aligning stage (see Input Files on
page 214). The file should conform to the GTF/GFF format specification and should list annotated
transcripts that match the reference genome being mapped against. The similar GFF3 format is
currently not supported.
DRAGEN can take the SJ.out.tab file (see SJ.out.tab on page 217) as an annotations file to help guide
the aligner in a two-pass mode of operation.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
58
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Sample Sex
Use the --sample-sex option to specify the sample sex on the command line. The information is passed
to all callers. The CNV caller contains a separate sex inference feature, but the specified --sample-sex
takes precedence over the inferred sex. The following example specifies sample sex using the --
sample-sex option:
--sample-sex MALE
--sample-sex FEMALE
If --sample-sex is not specified on the command line, the Ploidy Estimator runs by default to determine
the sex.
Stream Input Files
DRAGEN can stream input files directly from an AWS S3 bucket, or using HTTP presigned URLs. You do
not need to download the input files to a local disk prior to being processed. The files are streamed over
the network directly into the DRAGEN processor.
Input streaming is most beneficial for large input files. DRAGEN supports input streaming for BAMs and
compressed FASTQ files. For FASTQ files, input streaming can be used in all the configurations that
use single-end FASTQs, paired-end FASTQs, and FASTQ lists.
Input streaming is supported for the following use cases.
• Mapping/aligning of FASTQ and BAM.
• Germline and somatic small variant calling from BAM (without remapping).
For other file types that are significantly smaller in size, download them locally before running the
analysis.
Security and Permissions
To stream input files, you must have permission to access the remote files. The S3 object requires
AWS authentication and credentials. The authentication should already be set up on the instance you
are running, for example, via IAM policies. An HTTP URL most likely has a query string attached to it,
which provides the authentication credentials or necessary tokens to grant permission.
Examples
The following examples display possible methods to stream input files directly with DRAGEN.
Streaming FASTQ Input Using S3
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 s3://s3-bucket-name/path/to/object_1.fastq.gz \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
59
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
-2 s3://s3-bucket-name/path/to/object_2.fastq.gz \
--RGID object_ID \
--RGSM sample_name \
--output-directory /staging/examples/ \
--output-file-prefix streaming
Streaming FASTQ Input Using HTTP
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-1 https://bucket-name.amazonaws.com/path/to/object_1.fastq.gz?querystring
\
-2 https://bucket-name.amazonaws.com/path/to/object_2.fastq.gz?querystring
\
--RGID object_ID \
--RGSM sample_name \
--output-directory /staging/examples/ \
--output-file-prefix streaming
Streaming BAM Input Using S3
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b s3://s3-bucket-name/path/to/object_1.bam \
--output-directory /staging/examples/ \
--output-file-prefix streaming
Streaming BAM Input Using HTTP
dragen -f
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
-b https://bucket-name.amazonaws.com/path/to/object_1.bam?querystring \
--output-directory /staging/examples/ \
--output-file-prefix streaming
Autogenerated MD5SUM for BAM and CRAM Output Files
An MD5SUM file is generated automatically for BAM and CRAM output files. The MD5SUM file has the
same name as the output file with an .md5sum extension appended (eg, whole_genome_run_
123.bam.md5sum). The MD5SUM file is a single-line text file that contains the md5sum of the output
file, which exactly matches the output of the Linux md5sum command.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
60
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The MD5SUM calculation is performed as the output file is written, so there is no measurable
performance impact (compared to the Linux md5sum command, which can take several minutes for a
30x BAM).
Configuration Files
Command-line options can be stored in a configuration file.The location of the default configuration
file is /opt/edico/config/dragen-user-defaults.cfg. To specify a different file, use the --config-
file (-c) option.The configuration file used for a given run supplies the default settings for that run, any
of which can be overridden by command-line options.
The recommended approach is to use the dragen-user-defaults.cfg file as a template to create
default settings for different use cases. Copy dragen-user-defaults.cfg, rename the copy, and
then modify the new file for the specific use-case. As a best practice, put options that rarely change
into the configuration file and specify options that vary per run on the command-line.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
61
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN DNA Pipeline
Figure 2 DNA Pipeline for DRAGEN
DNA Mapping
Seed Density
The seed-density option controls how many (normally overlapping) primary seeds from each read the
mapper looks up in its hash table for exact matches. The maximum density value of 1.0 generates a
seed starting at every position in the read, ie, (L-K+1) K-base seeds from an L-base read.
Seed density must be between 0.0 and 1.0.Internally, an available seed pattern equal or close to the
requested density is selected. The most sparse pattern is one seed per 32 positions, or density
0.03125.
• Accuracy Considerations—Generally, denser seed lookup patterns improve mapping
accuracy.However, for long reads ( 50 bp+) and low sequening error rates, there is minimum
improvement beyond the default 50% seed lookup density..
• Speed Considerations—Denser seed lookup patterns generally slow down mapping, while more
sparse seed patterns speed up mapping.However, when the seed-mapping stage runs faster than
the aligning stage, a sparser seed pattern does not improve the mapper speed.
Relationship to Reference Seed Interval
Functionally, a denser or more sparse seed lookup pattern has an impact similar to a shorter or longer
reference seed interval (build hash table option --ht-ref-seed-interval).Populating 100% of reference
seed positions and looking up 50% of read seed positions has the same effect as populating 50% of
reference seed positions and looking up 100% of read seed positions. Either way, the expected density
of seed hits is 50%.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
62

More generally, the expected density of seed hits is the product of the reference seed density (the
inverse of the reference seed interval) and the seed lookup density. For example, if 50% of reference
seeds are populated and 33.3% (1/3) of read seed positions are looked up, then the expected seed hit
density should be 16.7% (1/6).
Local Analysis Software automatically adjusts theseed lookup pattern to make sure it does not
systematically miss the seed positions populated from the reference. For example, the mapper does
not look up seeds matching only odd positions in the reference when only even positions are populated
in the hash table, even if the reference seed interval is 2 and seed-density is 0.5.
Map Orientations
The --Mapper.map-orientations option is used in mapping reads for bisulfite methylation analysis.The
option is set automatically based on the value set for --methylation-protocol.
The --Mapper.map-orientations option can restrict the orientation of read mapping to only forward in
the reference genome or only reverse-complemented.The following are the valid values for --map-
orientations:
• 0—Either orientation (default).
• 1—Only forward mapping.
• 2—Only reverse-complemented mapping.
If mapping orientations are restricted and paired end reads are used, the expected pair orientation can
only be FR, not FF, or RF.
Seed Density Command-Line Options
Although Local Analysis Software primarily maps reads by finding exact reference matches to short
seeds, it can also map seeds differing from the reference by one nucleotide by also looking up single-
SNP edited seeds.Seed editing is usually not necessary with longer reads (100 bp+), because longer
reads have a high probability of containing at least one exact seed match.This is especially true when
paired ends are used, because a seed match from either mate can successfully align the
pair.However, seed editing can be useful to increase mapping accuracy for short single-ended reads,
with some cost in increased mapping time.The following options control seed editing:
Command-Line Option Name Configuration File Option Name
--Mappper.seed-density seed-density
--Mapper.edit-mode edit-mode
--Mapper.edit-seed-num edit-seed-num
--Mapper.edit-read-len edit-read-len
Table 3 Seed Editing Options
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
63
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Command-Line Option Name Configuration File Option Name
--Mapper.edit-chain-limit edit-chain-limit
edit-mode and edit-chain-limit
The edit-mode and edit-chain-limit options control when seed editing is used.The following four edit-
mode values are available:
Mode
Value
Description
0 No editing (default).
1 Chain length test.
2 Paired chain length test.
3 Full seed editing.
Edit mode 0 requires all seeds to match exactly. Mode 3 is the most expensive because every seed that
fails to match the reference exactly is edited.Modes 1 and 2 employ heuristics to look up edited seeds
only for reads most likely to be salvaged to accurate mapping.
The main heuristic in edit modes 1 and 2 is a seed chain length test.Exact seeds are mapped to the
reference in a first pass over a given read, and the matching seeds are grouped into chains of similarly
aligning seeds.If the longest seed chain in the read exceeds a threshold edit-chain-limit, the read does
not require seed editing, because there is already a mapping position.
Edit mode 1 triggers seed editing for a given read using the seed chain length test.If no seed chain
exceeds edit-chain-limit (including if no exact seeds match), then a second seed-mapping pass is
attempted using edited seeds.Edit mode 2 further optimizes the heuristic for paired-end reads.If
either mate has an exact seed chain longer than edit-chain-limit, then seed editing is disabled for the
pair because a rescue scan is likely to recover the mate alignment based on seed matches from one
read.Edit mode 2 is the same as mode 1 for single-end reads.
edit-seed-num and edit-read-len
For edit modes 1 and 2, when the heuristic triggers seed editing, these options control how many seed
positions are edited in the second pass over the read.Although exact seed mapping can use a densely
overlapping seed pattern, such as seeds starting at 50% or 100% of read positions, most of the value of
seed editing can be obtained by editing a much more sparse pattern of seeds, even a nonoverlapping
pattern.Generally, if a user application can afford to spend some additional amount of mapping time
on seed editing, a greater increase in mapping accuracy can be obtained for the same time cost by
editing seeds in sparse patterns for a large number of reads, than by editing seeds in dense patterns
for a small number of reads.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
64
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Whenever seed editing is triggered, these two options request edit-seed-num seed editing positions,
distributed evenly over the first edit-read-len bases of the read.For example, with 21-base seeds, edit-
seed-num=6 and edit-read-len=100, edited seeds can begin at offsets {0, 16, 32, 48, 64, 80} from the
5’ end, consecutive seeds overlapping by five bases.Because sequencing technologies often yield
better base qualities nearer the (5’) beginning of each read, this can focus seed editing where it is most
likely to succeed.When a particular read is shorter than edit-read-len, fewer seeds are edited.
Seed editing is more expensive when the reference seed interval (build hash table option --ht-ref-
seed-interval) is greater than 1.For edit modes 1 and 2, additional seed editing positions are
automatically generated to avoid missing the populated reference seed positions.For edit mode 3, the
time cost can increase dramatically because query seeds matching unpopulated reference positions
typically miss and trigger editing.
DNAAligning
Smith-Waterman Alignment Scoring Settings
The first stage of mapping is generates seeds from the read and looks for exact matches in the
reference genome. The seed match results are then refined by running full Smith-Waterman
alignments on the locations with the highest density of seed matches. The alignment algorithm
compares each position of the read against all candidate positions of the reference. These
comparisons correspond to a matrix of potential alignments between read and reference. For each of
the candidate alignment positions, Smith-Waterman generates scores that are used to evaluate
whether the best alignment passing through that matrix cell reaches it by a nucleotide match or
mismatch (diagonal movement), a deletion (horizontal movement), or an insertion (vertical movement).
A match between read and reference provides a bonus, on the score, and a mismatch or indel imposes
a penalty. The overall highest scoring path through the matrix is the alignment chosen.
The specific values chosen for scores in this algorithm indicate how to balance, for an alignment with
multiple possible interpretations, the possibility of an indel as opposed to one or more SNPs, or the
preference for an alignment without clipping. The default DRAGENscoring values are reasonable for
aligning moderate length reads to a whole human reference genome for variant calling applications.
But any set of Smith-Waterman scoring parameters represents an imprecise model of genomic
mutation and sequencing errors, and differently tuned alignment scoring values can be more
appropriate for some applications.
The following options control Smith-Waterman Alignment:
Command-Line Option
Name
Configuration File Option
Name
--Aligner.global global
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
65
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Command-Line Option
Name
Configuration File Option
Name
--Aligner.match-score match-score
--Aligner.match-n-score match-n-score
--Aligner.mismatch-pen mismatch-pen
--Aligner.gap-open-pen gap-open-pen
--Aligner.gap-ext-pen gap-ext-pen
--Aligner.unclip-score unclip-score
--Aligner.no-unclip-score no-unclip-score
--Aligner.aln-min-score aln-min-score
• global
The global option (value can be 0 or 1) controls whether alignment is forced to be end-to-end in the
read. When set to 1, alignments are always end-to-end, as in the Needleman-Wunsch global
alignment algorithm (although not end-to-end in the reference), and alignment scores can be
positive or negative. When set to 0, alignments can be clipped at either or both ends of the read, as
in the Smith-Waterman local alignment algorithm, and alignment scores are nonnegative.
Generally, global=0 is preferred for longer reads, so significant read segments after a break of
some kind (large indel, structural variant, chimeric read, and so forth) can be clipped without
severely decreasing the alignment score. Setting global=1 might not have the desired effect with
longer reads because insertions at or near the ends of a read can function as pseudoclipping. Also,
with global=0, multiple (chimeric) alignments can be reported when various portions of a read
match widely separated reference positions.
Using global=1 is sometimes preferable with short reads, which are unlikely to overlap structural
breaks, unable to support chimeric alignments, and are suspected of incorrect mapping if they
cannot align well end-to-end.
Consider using the unclip-score option, or increasing it, instead of setting global=1, to make a soft
preference for unclipped alignments.
• match-score
The match-score option is the score for a read nucleotide matching a reference nucleotide (A, C, G,
or T). Its value is an unsigned integer, from 0 to 15. match_score=0 can only be used when global=1.
A higher match score results in longer alignments, and fewer long insertions.
• match-2-score
The match-2-score option is the score for a read nucleotide matching a 2-base IUPAC-IUB code in
the reference (K, M, R, S, W, or Y). This option is a signed integer, ranging from -16 to 15.
• match-3-score
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
66
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The match-3-score option is the score for a read nucleotide matching a 3-base IUPAC-IUB code in
the reference (B, D, H, or V). This option is a signed integer, from -16 to 15.
• match-n-score
The match-n-score option is the score for a read nucleotide matching an N code in the reference.
This option is a signed integer, from -16 to 15.
• mismatch-pen
The mismatch-pen option is the penalty (negative score) for a read nucleotide mismatching any
reference nucleotide or IUPAC-IUB code (except ‘N’, which cannot mismatch). This option is an
unsigned integer, from 0 to 63. A higher mismatch penalty results in alignments with more
insertions, deletions, and clipping to avoid SNPs.
• gap-open-pen
The gap-open-pen option is the penalty (negative score) for opening a gap (ie, an insertion or
deletion). This value is only for a 0-base gap. It is always added to the gap length times gap-ext-
pen. This option is an unsigned integer, from 0 to 127. A higher gap open penalty causes fewer
insertions and deletions of any length in alignment CIGARs, with clipping or alignment through
SNPs used instead.
• gap-ext-pen
The gap-ext-pen option is the penalty (negative score) for extending a gap (ie, an insertion or
deletion) by one base. This option is an unsigned integer, from 0 to 15. A higher gap extension
penalty causes fewer long insertions and deletions in alignment CIGARs, with short indels, clipping,
or alignment through SNPs used instead.
• unclip-score
The unclip-score option is the score bonus for an alignment reaching the beginning or end of the
read. An end-to-end alignment receives twice this bonus. This option is an unsigned integer, from 0
to 127. A higher unclipped bonus causes alignment to reach the beginning and/or end of a read
more often, where this can be done without too many SNPs or indels.
A nonzero unclip-score is useful when global=0 to make a soft preference for unclipped
alignments. Unclipped bonuses have little effect on alignments when global=1, because end-to-
end alignments are forced anyway (although 2 × unclip-score does add to every alignment score
unless no-unclip-score = 1). Illuminarecommendsusing the default for unclip-score when global=1,
because some internal heuristics consider how local alignments would have been clipped.
For longer reads, setting unclip-score much higher than gap-open-pen can result in insertions at or
near one end of a read being utilized as pseudoclipping, as happens with global=1.
• no-unclip-score
The no-unclip-score option can be 0 or 1. The default is 1. When no-unclip-score is set to 1, any
unclipped bonus (unclip-score) contributing to an alignment is removed from the alignment score
before further processing, such as comparison with aln-min-score, comparison with other
alignment scores, and reporting in AS or XS tags. However, the unclipped bonus still affects the
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
67
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
best-scoring alignment found by Smith-Waterman alignment to a given reference segment, biasing
toward unclipped alignments.
If unclip-score > 0 causes a Smith-Waterman local alignment to extend out to one or both ends of
the read, the alignment score stays the same or increases if no-unclip-score=0, whereas it stays
the same or decreases if no-unclip-score=1.
The default, no-unclip-score=1, is recommended when global=1, because every alignment is end-
to-end, and there is no need to add the same bonus to every alignment.
When changing no-unclip-score, consider whether aln-min-score should be adjusted. When no-
unclip-score=0, unclipped bonuses are included in alignment scores compared to the aln-min-
score floor, so the subset of alignments filtered out by aln-min-score can change significantly with
no-unclip-score.
• aln-min-score
The aln-min-score option specifies a minimum acceptable alignment score. Any alignment results
scoring lower are discarded. Increasing or decreasing aln-min-score can reduce or increase the
percentage of reads mapped. This option is a signed integer (negative alignment scores are
possible with global=0).
aln-min-score also affects MAPQ estimates. The primary contributor to MAPQ calculation is the
difference between the best and second-best alignment scores, and aln-min-score serves as the
suboptimal alignment score if nothing higher was found except the best score. Therefore,
increasing aln-min-score can decrease reported MAPQ for some low-scoring alignments.
Paired-End Options
DRAGENcan process paired-end data passed via a pair of FASTQ files or in a single interleaved FASTQ
file. The hardware maps the two ends separately, and then determines a set of alignments that seem
most likely to form a pair in the expected orientation and having roughly the expected insert size. The
alignments for the two ends are evaluated for the quality of their pairing, with larger penalties for insert
sizes far from the expected size. The following options control processing of paired-end data:
• Reorientation
The pe-orientation option specifies the expected paired-end orientation. Only pairs with this
orientation can be flagged as proper pairs. Valid values are as follows:
• 0—FR (default)
• 1—RF
• 2—FF
• unpaired-pen
For paired end reads, best mapping positions are determined jointly for each pair, according to the
largest pair score found, considering the various combinations of alignments for each mate. A pair
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
68
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
score is the sum of the two alignment scores minus a pairing penalty, which estimates the
unlikelihood of insert lengths further from the mean insert than this aligned pair.
The unpaired-pen option specifies how much alignment pair scores should be penalized when the
two alignments are not in properly paired position or orientation. This option also serves as the
maximum pairing penalty for properly paired alignments with extreme insert lengths.
The unpaired-pen option is specified in Phred scale, according to its potential impact on MAPQ.
Internally, it is scaled into alignment score space based on Smith-Waterman scoring parameters.
• pe-max-penalty
The pe-max-penalty option limits how much the estimated MAPQ for one read can increase
because its mate aligned nearby. A paired alignment is never assigned MAPQ higher than the
MAPQ that it would have received mapping single-ended, plus this value. By default, pe-max-
penalty = mapq-max = 255, effectively disabling this limit.
The key difference between unpaired-pen and pe-max-penalty is that unpaired-pen affects
calculated pair scores and thus which alignments are selected and pe-max-penalty affects only
reported MAPQ for paired alignments.
Mean Insert Size Detection
When working with paired-end data, DRAGENchooses among the highest-quality alignments for the
two ends to try to choose likely pairs. To make this choice, DRAGEN uses a Gaussian statistical model
to evaluate the likelihood that a pair of alignments constitutes a pair. This model is based on the
intuition that a particular library prep tends to create fragments of roughly similar size, thus producing
pairs whose insert lengths cluster well around some mean insert length.
If you know the statistics of your library prep for an input file (and the file consists of a single read
group), you can specify the characteristics of the insert-length distribution: mean, standard deviation,
and three quartiles. These characteristics can be specified with the Aligner.pe-stat-mean-insert,
Alinger.pe-stat-stddev-insert, Aligner.pe-stat-quartiles-insert, and Aligner.pe-stat-mean-read-
lenoptions.However, it is typically preferable to allow DRAGEN to detect these characteristics
automatically.
To enable automatic sampling of the insert-length distribution, set --enable-sampling to true. When
the software starts execution, it runs a sample of up to 100,000 pairs through the aligner, calculates
the distribution, and then uses the resulting statistics for evaluating all pairs in the input set.
The DRAGEN host software reports the statistics in its stdout log, as follows:
Final paired-end statistics detected for read group 0, based on 79935 high
quality pairs for FR orientation
Quartiles (25 50 75) = 398 410 421
Mean = 410.151
Standard deviation = 14.6773
Boundaries for mean and standard deviation: low = 352, high = 467
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
69
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Boundaries for proper pairs: low = 329, high = 490
NOTE: DRAGEN's insert estimates include corrections for
clipping (so they are no identical to TLEN)
The insert length distribution for each sample is written to fragment_length_hist.csv. Each sample
starts with the following lines
#Sample: sample name
FragmentLength,Count
These lines are followed by the histogram.
When the number of sample pairs is very small, there is not enough information to characterize the
distribution with high confidence. In this case, DRAGEN applies default statistics that specify a very
wide insert distribution, which tends to admit pairs of alignments as proper pairs, even if they may lie
tens of thousands of bases apart. In this situation, DRAGEN outputs a message, as follows:
WARNING: Less than 28 high quality pairs found - standard deviation is
calculated from the small samples formula
The small samples formula calculates standard deviation as follows:
if samples < 3 then
standard deviation = 10000
else if samples < 28 then
standard deviation = 25 * (standard deviation + 1) / (samples – 2)
end if
if standard deviation < 12 then
standard deviation = 12
end if
The default model is “standard deviation = 10000”. If the first 100,000 reads are unmapped or if all pairs
are improper pairs, then the standard deviation is set to 10,000 and the mean and quartiles are set to 0.
The minimum value for standard deviation is 12, which is independent of the number of samples.
For RNA-Seq data, the insert size distribution is not normal due to pairs containing introns. The
DRAGENN software estimates the distribution using a kernel density estimator to fit a long tail to the
samples. This estimate leads to a more accurate mean and standard deviation for RNA-Seq data and
proper pairing.
DRAGEN writes detected paired-end stats into a tab-delimited log file in the output directory called
.insert-stats.tab. This file contains the statistical distribution of detected insert sizes for each
read group, including quartiles, mean, standard deviation, minimum, and maximum. The information
matches the standard-out report above. Additionally, the log file includes the minimum and maximum
insert limits that DRAGEN applied for rescue scans.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
70
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Rescue Scans
For paired-end reads, where a seed hit is found for one mate but not the other, rescue scans hunt for
missing mate alignments within a rescue radius of the mean insert length.DRAGEN host software sets
the default rescue radius to 2.5 standard deviations of the empirical insert distribution.In cases where
the insert standard deviation is large compared to the read length, the rescue radius is restricted to
limit mapping slowdowns. In this case, a warning message is displayed, as follows:
Rescue radius = 220
Effective rescue sigmas = 0.5
WARNING: Default rescue sigmas value of 2.5 was overridden by host software!
The user may wish to set rescue sigmas value explicitly with --Aligner.rescue-sigmas
Although the user can ignore this warning, or specify an intermediate rescue radius to maintain
mapping speed, it is recommended to use 2.5 sigmas for the rescue radius to maintain mapping
sensitivity.To disable rescue scanning, set max-rescues to 0.
Output Options
DRAGEN can track multiple independent alignments for each read. These alignments include the
optimal (primary) one, as well as those mapping different subsegments of the read,
(chimeric/supplementary), and sub-optimal (secondary) mappings of the read to different areas of the
reference.
For DNA alignment by default, DRAGEN can emit one primary alignment for each read, up to three
chimeric alignments (Aliger.supp-aligns=3), and no secondary alignments (Aligner.sec-aligns=0). The
maximum user-specified value for supp-aligns or sec-aligns is 30. The maximum total number of
emitted alignments per read is 31. If the sum of supp-aligns and sec-aligns is greater than 30, then
chimeric alignments are tracked with higher priority.
Use the following configuration options to control how many of each type of alignment to include in
DRAGEN output.
• mapq-max
The mapq-max option specifies a ceiling on the estimated MAPQ that can be reported for any
alignment, from 0 to 255. If the calculated MAPQ is higher, this value is reported instead. The
default is 60.
• supp-aligns, sec-aligns
The supp-aligns and sec-aligns options restrict the maximum number of supplementary (ie,
chimeric and SAM FLAG 0x800) alignments and secondary (ie, suboptimal and SAM FLAG 0x100)
alignments, respectively, that can be reported for each read.
A maximum of 31 alignments are reported for any read total, including primary, supplementary, and
secondary. Therefore, supp-aligns and sec-aligns each range from 0–30. Supplementary
alignments are tracked and output with higher priority than secondary ones.
High settings for these two options impact speed so it is advisable to increase only as needed.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
71
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• sec-phred-delta
The sec-phred-delta option controls which secondary alignments are emitted based on the
alignment score relative to the primary reported alignment. Only secondary alignments with
likelihood within this Phred value of the primary are reported.
• sec-aligns-hard
The sec-aligns-hard option suppresses the output of all secondary alignments if there are more
secondary alignments than can be emitted. Set sec-aligns-hard to 1 to force the read to be
unmapped when not all secondary alignments can be output.
• supp-as-sec
When the supp-as-sec option is set to 1, then supplementary (chimeric) alignments are reported
with SAM FLAG 0x100 instead of 0x800. The default is 0. The supp-as-sec option provides
compatibility with tools that do not support FLAG 0x800.
• hard-clips
The hard-clips option is used as a field of 3 bits, with values ranging from 0 to 7. The bits specify
alignments, as follows:
– Bit 0—primary alignments
– Bit 1—supplementary alignments
– Bit 2—secondary alignments
Each bit determines whether local alignments of that type are reported with hard clipping (1) or soft
clipping (0). The default is 6, meaning primary alignments use soft clipping and supplementary and
secondary alignments use hard clipping.
DRAGEN Graph Mapper
To improve variant calling accuracy in segmental duplications and other regions difficult to map with
Illumina reads, you can use the graph mapper in DRAGEN. The graph-based method uses alt-aware
mapping for population haplotypes stitched into the reference with known alignments to establish
alternate graph paths that reads could seed-map and align to. The graph mapper reduces mapping
ambiguity because reads that contain population variants are attracted to the specific regions where
the variants are observed. Graph mapper is supported only for the hg38 reference.
DRAGEN augments the FASTA reference with around 900,000 short alternate contigs derived from
population haplotypes of phased variants to evolve the FASTA reference to a graph reference. The
DRAGEN mapper’s alt-aware capabilities are used to project reads that match the population
haplotypes to corresponding primary assembly alignments with a precise lift-over alignment.
When given a set of population variants (VCF) or haplotypes, the FASTA modification is categorized in
the following two types:
• Alternate contigs represent population haplotypes. Alt-contigs can have a single variant or a
combination of nearby phased variants.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
72
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Ambiguous codes (IUPAC codes) to represent SNPs . To improve alignment, edit the reference
FASTA with isolated population SNPs.
DRAGEN graph mapper requires hash tables that are built using a FASTA containing population
alternate contigs, corresponding lift-over SAM and unphased SNP VCF files are specified. The
following is an example DRAGEN command line to build a graph-based hash table:
dragen --build-hash-table true \
--ht-build-rna-hashtable true --enable-cnv true \
--ht-reference hg38.fa \
--output-directory /tmp/ --ht-num-threads 40 \
--ht-alt-liftover /opt/edico/liftover/bwa-kit_hs38DH_liftover.sam \
--ht-pop-alt-contigs /opt/edico/liftover/pop_altContig.fa.gz \
--ht-pop-alt-liftover /opt/edico/liftover/pop_liftover.sam.gz \
--ht-pop-snps /opt/edico/liftover/pop_snps.vcf.gz
Read Trimming
DRAGEN can remove artifacts from reads using hardware accelerated read trimming. Hardware
accelerated read trimming is available on U200 and AWS systems, as part of the DRAGEN mapper and
adds no additional run time. To enable the trimmer in normal mode, use --read-trimmers. To enable
the trimmer in soft mode, use --soft-read-trimmers.
In normal trimming mode, potential artifacts are removed from input reads. Reads which are trimmed
to fewer than 20 bases are filtered and replaced with a dummy read consisting of 10 N bases. In
addition, the filtered reads have 0x200 SAM flag set.
DRAGEN contains a novel lossless soft-trimming mode. In soft-trimming mode, reads are mapped as
though they had been trimmed, but no bases are removed. The intention of soft trimming is to
suppress systematic mismapping of reads containing trimmable artifacts, such as Poly-G artifacts,
from getting mapped to reference G homopolymers or adapter sequences getting mapped to matching
reference loci, without actually losing the trimmed bases in aligned output. Soft trimming for Poly-G
artifacts is enabled by default on supported systems.
Read Trimming Metrics
The trimmer generates a metrics file titled <output prefix>.trimmer_metrics.csv. Metrics are
available on an aggregate level over all input data. The metrics units are in reads or bases.
• Total input reads—Total number of reads in the input files.
• Total input bases—Total number of bases in the input reads.
• Total input bases R1—Total number of bases in R1 reads.
• Total input bases R2—Total number of bases in R2 reads.
• Average input read length—Total number of input bases divided by the number of input reads.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
73
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Total trimmed reads—Total number of reads trimmed by at least one base, not including soft-
trimming.
• Total trimmed bases—Total number of bases trimmed, not including soft-trimming.
• Average bases trimmed per read—The number of trimmed bases divided by the number of input
reads.
• Average bases trimmed per trimmed read—The number of trimmed bases divided by the number
of trimmed reads.
• Remaining poly-G K-mers R1 3prime—The number of R1 3' read ends that contain likely Poly-G
artifacts after trimming.
• Remaining poly-G K-mers R2 3prime—The number of R2 3' read ends thatcontain likely Poly-G
artifacts after trimming.
• Poly-G trimmed reads—The number of reads with at least one base trimmed during Poly-G artifact
trimming. This metric is reported for both mates (R1 and R2) and the filtering status (unfiltered and
filtered) of the trimmed read. The metric includes reads which were trimmed during soft-trimming.
• Poly-G trimmed bases—The number of bases trimmed during Poly-G artifact trimming. This metric
is reported for both mates (R1 and R2) and the filtering status (unfiltered and filtered) of the
trimmed read. The metric includes bases from reads which were trimmed during soft-trimming.
• Total filtered reads—The number of reads that were filtered out during trimming.
• Reads filtered for minimum read length R1—The number of R1 reads that were filtered due to
being trimmed below the minimum read length.
• Reads filtered for minimum read length R2—The number of R2 reads that were filtered due to
being trimmed below the minimum read length.
Poly-G Trimming
Poly-G artifacts appear on two-channel sequencing system when the dark base G is called after
synthesis has terminated. This results in calling several erroneous high-confidence G bases on the
ends of affected reads. For contaminated samples, a large number of affected reads can be mapped to
reference regions with high G content. This can cause problems for processing downstream.
Read Trimming Settings
Use the following options to configure read trimming:
• --read-trimmers—To enable Poly-G trimming, set to polyg. To disable Poly-G trimming, set to
none. During mapping, artifacts are removed from all reads. Reads are mapped accordingly.
• --soft-read-trimmers—To enable soft Poly-G trimming, set to polyg. To disable Poly-G trimming,
set to none. During mapping, reads are aligned as if they had been trimmed. No bases are removed
from the reads. Soft-trimming is enabled by default.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
74
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN FastQC
DRAGEN's FastQC module is a tool for calculating common metrics used for quality control of high-
throughput sequencing data. The tool is modeled after the metrics generated by Babraham Institute's
FastQC tool.
The metrics are generated automatically on all DRAGEN map-align workflows with no additional run
time and output in a CSV format file called <PREFIX>.fastqc_metrics.csv.
For users only interested in sample QC or would like to obtain FastQC results only, DRAGEN provides a
mode to generate the fastqc_metrics.csv file directly.
By default DRAGEN FastQC and read-trimming are run as preprocessing steps to standard sequence
alignment workflows. If DNA alignment is not needed or if QC results are needed more quickly, the
mapping and BAM output portions of the workflow can be disabled. The workflow only outputs key
metric files and runs ~70% faster. This option is available on the command-line by entering --fastqc-
only=true after the DRAGEN command.
Metric Granularity
It is not possible due to memory constraints to guarantee single-base resolution for all metrics.
DRAGEN provides an algorithmic solution for binning via --fastqc-granularity. DRAGEN allocates
256 bins in memory for each size or position-based metric. The granularity value of 4–7 inclusive can be
used to determine the bin size. High values use smaller bins for greater resolution. Lower values can be
used to create larger bins for larger read-lengths.
Granularity
Single Base Resolution
(bp)
Resolution at 150 (bp)
Recommended Read-Lengths
(bp)
7 [default] 1–255 1 < 256
6 1–128 2 ≥ 256 and < 507
5 1–64 4 ≥ 507 and < 4031
4 1–32 8 ≥ 4031
Adapter and Kmer Sequence Files
To include metrics for adapter or other sequence content, DRAGEN FastQC needs to be provided with
the desired sequences in FASTA format. DRAGEN provides two options for this purpose, --fastqc-
adapter-file for adapter sequences and --fastqc-kmer-file for any additional kmers of interest
so that users can add sequences of interest without changing the expected adapter results.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
75
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
DRAGEN FastQC can accept up to a combined total of 16 adapters and kmer sequences. Each
sequence can be a maximum of 12 bp in length. By default, DRAGEN uses the adapter file located at
/opt/edico/config/adapter_sequences.fasta. The file contains the following same adapter
sequences as Babraham's FastQC v 0.11.10 and later.
• Illumina Universal Adapter—AGATCGGAAGAG
• Illumina Small RNA 3' Adapter—TGGAATTCTCGG
• Illumina Small RNA 5' Adapter—GATCGTCGGACT
• Nextera Transposase Sequence—CTGTCTCTTATA
FastQC Metrics Output
The FastQC metrics are output to a CSV file format in the run output directory called
<PREFIX>.fastqc_metrics.csv.
The reported metrics are broken down into eight sections by metric type. Each section is broken down
further into separate rows by either the length, position, or other relevant categorical variables. The
following are the metric sections.
• Read Mean Quality—Total number of reads. Each average Phred-scale quality value is rounded to
the nearest integer.
• Positional Base Mean Quality—Average Phred-scale quality value of bases with a specific
nucleotide and at a given location in the read. Locations are listed first and can be either specific
positions or ranges. The nucleotide is listed second and can be A, C, G, or T. N or ambiguous bases
are assumed to have the system default value, usually QV2.
• Positional Base Content—Number of bases of each specific nucleotide at given locations in the
read. Locations are given first and can be either specific positions or ranges. The nucleotide is listed
second and can be A, C, G, T, N.
• Read Lengths— Total number of reads with each observed length. Lengths can be either specific
sizes or ranges, depending on the settings specified using --fastqc-granularity.
• Read GC Content—Total number of reads with each GC content percentile between 0 % and 100
%.
• Read GC Content Quality—Average Phred-scale read mean quality for reads with each GC content
percentile between 0% and 100%.
• Sequence Positions—Number of times an adapter or other kmer sequence is found, starting at a
given position in the input reads. Sequences are listed first in the metric description in quotes.
Locations are listed second and can be either specific positions or ranges.
• Positional Quality—Phred-scale quality value for bases at a given location and a given quantile of
the distribution. Locations are listed first and can be either specific positions or ranges. Quantiles
are listed second and can be any whole integer 0–100.
The following are examples rows from each section.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
76
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Section Mate Metric Value
READ MEAN QUALITY Read1 Q38 Reads 965377
...
POSITIONAL BASE MEAN
QUALITY
Read1 ReadPos 145-152 T Average
Quality
34.49
POSITIONAL BASE MEAN
QUALITY
Read1 ReadPos 150 T Average Quality 34.44
POSITIONAL BASE MEAN
QUALITY
Read1 ReadPos 256+ T Average Quality 36.99
...
POSITIONAL BASE CONTENT Read1 ReadPos 145-152 A Bases 113362306
POSITIONAL BASE CONTENT Read1 ReadPos 150 A Bases 14300589
POSITIONAL BASE CONTENT Read1 ReadPos 256+ A Bases 13249068
...
READ LENGTHS Read1 150bp Length Reads 77304421
READ LENGTHS Read1 144-151bp Length Reads 77304421
READ LENGTHS Read1 >=255bp Length Reads 1000000
...
READ GC CONTENT Read1 50% GC Reads 140878674373
...
READ GC CONTENT QUALITY Read1 50% GC Reads Average Quality 36.20
...
SEQUENCE POSITIONS Read1 'AGATCGGAAGAG' 137bp Starts 20
SEQUENCE POSITIONS Read1 'AGATCGGAAGAG' 137-144bp
Starts
23
...
POSITIONAL QUALITY Read1 ReadPos 150 50% Quantile QV 37
POSITIONAL QUALITY Read1 ReadPos 145-152 50% Quantile QV 37
...
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
77
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
ALT-Aware Mapping
The GRCh38 human reference contains many more alternate haplotypes (ALT contigs) than previous
versions of the reference. Generally, including ALT contigs in the mapping reference improves mapping
and variant calling specificity, because misalignments are eliminated for reads matching an ALT contig
but scoring poorly against the primary assembly. However, mapping with GRCh38’s ALT contigs
without special treatment can substantially degrade variant calling sensitivity in corresponding regions,
because many reads align equally well to an ALT contig and to the corresponding position in the primary
assembly. DRAGEN ALT-aware mapping eliminates this issue, and instead obtains both sensitivity and
specificity improvements from ALT contigs.
ALT-aware mapping requires hash tables that are built with ALT liftover alignments specified (see ALT-
Aware Hash Tables on page 13). If a hash table built with liftover alignments is provided, DRAGEN
automatically runs with ALT-aware mapping. Set the --alt-aware option to false to disable ALT-Aware
mapping with a liftover reference.
DRAGEN requires ALT-Aware hash tables for any hg19 or GRCh38 reference where ALT contigs are
detected. To disable this requirement in DRAGEN, set the --ht-alt-aware-validate option to false.
When ALT-aware mapping is enabled, the mapper and aligner are aware of the liftover relationship
between ALT contig positions and corresponding primary assembly positions. Seed matches within ALT
contigs are used to obtain corresponding primary assembly alignments, even if the latter score poorly.
Liftover groups are formed, each containing a primary assembly alignment candidate, and zero or more
ALT alignment candidates that lift to the same location. Each liftover group is scored according to its
best-matching alignments, taking properly paired alignments into account. The winning liftover group
provides its primary assembly representative as the primary output alignment, with MAPQ calculated
based on the score difference to the second-best liftover group. Emitting primary alignments within the
primary assembly maintains normal aligned coverage and facilitates variant calling there. If the --
Aligner.en-alt-hap-aln option is set to 1 and --Aligner.supp-aligns is greater than 0, then corresponding
alternate haplotype alignments can also be output, flagged as supplementary alignments.
The following is a comparison of alternative options for dealing with alternate haplotypes.
• Mapping without ALT contigs in the reference:
– False-positive variant calls result when reads matching an alternate haplotype misalign
somewhere else.
– Poor mapping and variant calling sensitivity where reads matching an ALT contig differ greatly
from the primary assembly.
• Mapping with ALT contigs but no ALT awareness:
– False-positive variant calls from misaligned reads matching ALT contigs are eliminated.
– Low or zero aligned coverage in primary assembly regions covered by alternate haplotypes, due
to some reads mapping to ALT contigs.
– Low or zero MAPQ in regions covered by alternate haplotypes, where they are similar or
identical to the primary assembly.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
78
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
– Variant calling sensitivity is dramatically reduced throughout regions covered by alternate
haplotypes.
• Mapping with ALT contigs and alt awareness:
– False-positive variant calls from misaligned reads matching ALT contigs are eliminated.
– Normal aligned coverage in regions covered by alternate haplotypes because primary
alignments are to the primary assembly.
– Normal MAPQs are assigned because alignment candidates within a liftover group are not
considered in competition.
– Good mapping and variant calling sensitivity where reads matching an ALT contig differ greatly
from the primary assembly.
Sorting
The map/align system produces a BAM file sorted by reference sequence and position by
default.Creating this BAMfile typically eliminates the requirement to run samtools sort or any
equivalent postprocessing command. The --enable-sort option can be used to enable or disable
creation of the BAMfile, as follows:
• To enable, set to true.
• To disable, set to false.
On the reference hardware system, running with sort enabled increases run time for a 30x full genome
by about 6–7 minutes.
Duplicate Marking
Marking or removing duplicate aligned reads is a common best practice in whole-genome sequencing.
Not doing so can bias variant calling and lead to incorrect results.
The DRAGENsystem can mark or remove duplicate reads, and produce a BAM file with duplicates
either marked in the FLAG field or entirely removed.
Algorithm
The DRAGEN duplicate-marking algorithm is modeled on the Picard toolkit MarkDuplicates feature. All
the aligned reads are grouped into subsets in which all the members of each subset are potential
duplicates.
For two pairs to be duplicates, they must have the following:
• Identical alignment coordinates (position adjusted for soft- or hard-clips from the CIGAR) at both
ends.
• Identical orientations (direction of the two ends, with the left-most coordinate being first).
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
79
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

In addition, an unpaired read may be marked as a duplicate if it has identical coordinate and orientation
with either end of any other read, whether paired or not.
Unmapped or read pairs are never marked as duplicates.
When DRAGEN identifies a group of duplicates, it picks one as the best of the group, and marks the
others with the BAM PCR or optical duplicate flag (0x400, or decimal 1024). For this comparison,
duplicates are scored based on the average sequence Phred quality. Pairs receive the sum of the
scores of both ends, while unpaired reads get the score of the one mapped end. The score is intended
to preserve the reads with the highest-quality base calls.
If two reads (or pairs) have exactly matching quality scores, DRAGEN breaks the tie by choosing the
pair with the higher alignment score. If there are multiple pairs that also tie on this attribute, then
DRAGENchooses a pairarbitrarily.
The score for an unpaired read R is the average Phred quality score per base, calculated as follows:
Where:
• R is a BAM record
• QUAL is its array of Phred quality scores
• dedup-min-qual is a DRAGEN configuration option with default value of 15.
For a pair, the score is the sum of the scores for the two ends.
This score is stored as a one-byte number, with values rounded down to the nearest one-quarter. This
rounding may lead to different duplicate marks from those chosen by Picard, but because the reads
were very close in quality this has negligible impact on variant calling results.
Limitations
The limitations to DRAGEN duplicate marking implementation are as follows:
• When there are two duplicate reads or pairs with very close Phred sequence quality scores,
DRAGEN might choose a different winner from that chosen by Picard.These differences have
negligible impact on variant calling results.
• If using a single FASTQ file as input, DRAGEN accepts only a single library ID as a command-line
argument (PGLB). For this reason, the FASTQ inputs to the system must be already separated by
library ID. Library ID cannot be used as a criterion for distinguishing non-duplicates.
Settings
The following options can be used to configure duplicate marking in DRAGEN:
• --enable-duplicate-marking
Set to true to enable duplicate marking.When --enable-duplicate-marking is enabled, the output is
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
80
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
sorted, regardless of the value of the enable-sort option.
• --remove-duplicates
Set to true to suppress the output of duplicate records.If set to false, set the 0x400 flag in the
FLAG field of duplicate BAM records. When --remove-duplicates is enabled, then enable-duplicate-
marking is forced to enabled as well.
• --dedup-min-qual
Specifies the Phred quality score below which a base should be excluded from the quality score
calculation used for choosing among duplicate reads.
Small Variant Calling
The DRAGEN Small Variant Caller is a high-speed haplotype caller implemented with a hybrid of
hardware and software. The caller performs localized de novo assembly in regions of interest to
generate candidate haplotypes and then performs read likelihood calculations using a hidden Markov
model (HMM).
Variant calling is disabled by default. You can enable variant calling by setting the --enable-variant-
caller option to true.
The Variant Caller Algorithm
The DRAGEN Haplotype Caller performs the following steps:
• Active Region Identification—Identifies areas where multiple reads disagree with the reference
are identified, and selects windows around them (active regions) for processing.
• Localized Haplotype Assembly— For each active region, assembles all overlapping reads in each
active region into a de Bruijn graph (DBG). A DBG is a directed graph based on overlapping K-mers
(length K sub-sequences) in each read or multiple reads. When all reads are identical, the DBG is
linear. Where there are differences, the graph forms bubbles of multiple paths diverging and
rejoining. If the local sequence is too repetitive and K is too small, cycles can form, which invalidate
the graph. Values of K=10 and 25 are tried by default. If those values produce an invalid graph, then
additional values of K = 35, 45, 55, 65 are tried until a cycle-free graph is obtained. From this cycle-
free DBG, every possible path is extracted to produce a complete list of candidate haplotypes, ie,
hypotheses for what the true DNA sequence may be on at least one strand.
• Haplotype Alignment—Uses the Smith-Waterman algorithm to align each extracted haplotype to
the reference genome to determine what variations from the reference it implies.
• Read Likelihood Calculation—Tests each read against each haplotype, to estimate a probability of
observing the read assuming the haplotype was the true original DNA sampled. This calculation is
performed by evaluating a pair hidden Markov model (HMM), which accounts for the various
possible ways the haplotype might have been modified by PCR or sequencing errors into the read
observed. The HMM evaluation uses a dynamic programming method to calculate the total
probability of any series of Markov state transitions arriving at the observed read.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
81
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• Genotyping—Forms the possible diploid combinations of variant events from the candidate
haplotypes and, for each combination, calculates the conditional probability of observing the entire
read pileup. Calculations use the constituent probabilities of observing each read, given each
haplotype from the pair HMM evaluation. These calculations feed into the Bayesian formula to
calculate a likelihood that each genotype is the truth, given the entire read pileup observed.
Genotypes with maximum likelihood are reported.
Filter BAM Input
Information in the IGV BAM pileup differs from the INFO/DP and FORMAT/DP in the VCF/gVCF as a
result of filtering steps applied throughout variant calling. There are four filters used to exclude reads
from the genotyping calculations. The following figure summarizes the four filters.
• Filter 1 filters out the following reads from the IGV BAM input:
– Duplicate reads.
– Reads with MAPQ=0.
– Soft-clipped bases. DRAGEN filters out soft-clipped bases only when calculating coverage
reports.
– [Somatic] Reads with MAPQ < vc-min-tumor-read-qual, where vc-min-tumor-read-qual >1.
• Filter 2 trims bases with BQ < 10 and filters out the following reads:
– Unmapped reads.
– Secondary reads.
– Reads with bad cigars.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
82
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Filter 3 occurs after down-sampling and HMM. Filter 3 filters out the following reads:
– Reads that are badly mated. A badly mated read is a read where the pair is mapped to two
different reference contigs.
– Disqualified reads. Reads are disqualified if their HMM score is below a threshold.
• Filter 4 occurs after the genotyper runs. The genotyper adds annotation information to the
FORMAT field. Filter 4 filters out reads that are not informative. For example, if the HMM scores of
the read against two different haplotypes are almost equal, the read is filtered out because it does
not provide enough information to distinguish which of the two haplotypes are more likely.
INFO/DP includes both informative and non-informative reads.
FORMAT/AD and FORMAT/DP include only informative reads.
Variant Caller Options
The following options control the variant caller stage of the DRAGEN host software.
• --enable-variant-caller
Set --enable-variant-caller to true to enable the variant caller stage for the DRAGEN pipeline.
• --vc-target-bed
This is an optional command line input that restricts processing of the small variant caller, target
bed related coverage, and callability metrics to regions specified in a BED file. The BED file is a text
file containing at least three tab-delimited columns. The first three columns are chromosome, start
position, and end position, respectively. The positions are zero-based. For example:
For example:
# header information
chr11 0 246920
chr11 255660 255661
• --vc-target-bed-padding
This is an optional command line input that can be used to pad all of the target BED regions with the
specified value. For example, if a BED region is 1:1000-2000 and a padding value of 100 is used, it is
equivalent to using a BED region of 1:900-2100 and a padding value of 0. Any padding added to --
vc-target-bed-padding is used by the small variant caller and by the target bed coverage/callability
reports. The default padding is 0.
• --vc-sample-name
The --vc-sample-name option is deprecated. In end-to-end germline mode (with FASTQ input), the
variant caller uses the RGSM value as the sample name. In end-to-end somatic mode, --RGSM-
tumor can be used to specify the sample name of the tumor sample. In stand-alone mode (with
BAM input), the variant caller uses the RGSM value from the BAM header as the sample name. In
somatic mode, the RGSM value from a tumor BAM is as the tumor sample name.
• --vc-target-coverage
The --vc-target-coverage option specifies the target coverage for down-sampling. The default
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
83
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
value is 500 for germline mode and 50 for somatic mode.
• --vc-enable-gatk-acceleration
If --vc-enable-gatk-acceleration is set to true, the variant caller runs in GATK mode (concordant
with GATK 3.7 in germline mode and GATK 4.0 in somatic mode).
• --vc-remove-all-soft-clips
If --vc-remove-all-soft-clips is set to true, the variant caller does not use soft clips of reads to
determine variants.
• --vc-decoy-contigs
The --vc-decoy-contigs option specifies a comma-separated list of contigs to skip during variant
calling. This option can be set in the configuration file.
• --vc-enable-decoy-contigs
If --vc-enable-decoy-contigs is set to true, variant calls on the decoy contigs are enabled. The
default value is false.
• --vc-enable-phasing
The –vc-enable-phasing option enables variants to be phased when possible. The default value is
true.
• --vc-enable-phasing
The –vc-enable-phasing option enables variants to be phased when possible. The default value is
true.
• --vc-enable-vcf-output
The –vc-enable-vcf-output option enables VCF file output during a gVCF run. The default value is
false.
Down-sampling Options for Small Variant Calling
The options for down-sampling reads in the small variant calling pipeline are as follows:
• --vc-target-coverage specifies the maximum number of reads with a start position overlapping any
given position.
• --vc-max-reads-per-active-region specifies the maximum number of reads covering a given active
region.
• --vc-max-reads-per-raw-region specifies the maximum number of reads covering a given raw
region.
• --vc-min-reads-per-start-pos specifies the minimum number of reads with a start position
overlapping any given position.
For mitochondrial small variant calling, the down-sampling options can be set separately because the
mitochondrial contig contains a higher depth than the rest of the contigs in a WGS dataset. The
following are the down-sampling options for the mitochondrial contig.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
84
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• --vc-target-coverage-mito
• --vc-max-reads-per-active-region-mito
• --vc-max-reads-per-raw-region-mito
The target coverage and max/min reads in raw/active region options are not directly related and could
be triggered independently.
The target coverage option runs first and is meant to limit the number of reads that share the same
start position at any given position. It is not a limit on the total coverage at a given position.
The following are the default down-sampling values for each small variant calling mode.
Mode Down-sampling Option
Default
Value
Germline --vc-target-coverage 500
Germline --vc-max-reads-per-active-region 10000
Germline --vc-max-reads-per-raw-region 30000
Somatic --vc-target-coverage 50
Somatic --vc-max-reads-per-active-region 10000
Somatic --vc-max-reads-per-raw-region 30000
High Coverage --vc-target-coverage 100000
High Coverage --vc-max-reads-per-active-region 200000
High Coverage --vc-max-reads-per-raw-region 200000
Mitochondrial --vc-target-coverage-mito 40000
Mitochondrial --vc-max-reads-per-active-region-mito 40000
Mitochondrial --vc-max-reads-per-raw-region-mito 40000
The following example that shows that the DP reported in a variant record can far exceed the --vc-
target-coverage default value of 500 in Germline mode:
For example, assume the default value of --vc-target-coverage is 500. If there are 400 reads starting
at position 1, another 400 starting at position 2, and another 400 starting at position 3, the target
coverage option is not triggered (because 400 < 500). If there is a variant at position 4, its reported
depth could be as high as 1200. This example shows that the DP reported in a variant record can far
exceed the --vc-target-coverage value.
After the target coverage step, the maximum number of reads that share the same position is 500 (if --
vc-target-coverage is set to 500).
The next down-sampling step is to apply the --vc-max-reads-per-raw-region and --vc-max-reads-
per-active-region limits. In this step, the maximum number of reads that share the same position can
be further reduced from the 500 maximum value from the first step. These options are used to limit the
total number of reads in an entire region using a leveling down-sampling method.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
85
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The down-sampling mechanism scans each start position from the start boundary of the region and
discards one read from that position, then moves on to the next position, until the total number of reads
falls below the threshold. It can potentially take several passes across the entire region for the total
number of reads in the entire region to fall below the threshold. After the threshold is met, the down-
sampling step is stopped regardless of which position was considered last in the region.
If the number of reads at any position with same start position is equal to or lower than the --vc-min-
reads-per-start-pos, that position is skipped to ensure that there is always at least a minimum number
of reads (set to --vc-min-reads-per-start-pos) at any start position.
When down-sampling occurs, the choice of which reads to keep or remove is somewhat random.
However, the random number generator is seeded to a default value to ensure that it produces the
same set of values in each run. This ensures exactly reproducible results, which means there is no run
to run variation when using the same input data.
Down-sampling Options for Mitochondrial Small Variant Calling
The options for down-sampling reads in the small variant calling pipeline for the mitochondrial contig
are as follows:
• --vc-target-coverage-mito specifies the maximum number of reads with a start position
overlapping any given position.
• --vc-max-reads-per-active-region-mito specifies the maximum number of reads covering a given
active region.
• --vc-max-reads-per-raw-region-mito specifies the maximum number of reads covering a given
raw region.
The default value for all three options is 40000. Their function is the same as described in Down-
sampling Options for Small Variant Calling on page 84. You can set the down-sampling options for the
mitochondrial contig separately from the rest of the contigs because the mitochondrial contig has much
higher depth than the rest of the contigs in a WGS dataset.
Phasing and Phased Variants
DRAGEN supports output of phased variant records in the germline VCF and gVCF file. When two or
more variants are phased together, the phasing information is encoded in a sample-level annotation,
FORMAT/PS, that identifies which set the phased variant is in. The value in the field in an integer
representing the position of the first phased variant in the set. All records in the same contig with
matching PS values belong to the same set.
##FORMAT=<ID=PS,Number=1,Type=Integer,Description="Physical phasing ID
information, where each unique ID within a given sample (but not across
samples) connects records within a phasing group">
The following is an example of a DRAGEN single sample gVCF, where two SNPs are phased together.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
86
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
chr1 1947645 . C T,<NON_REF> 48.44 PASS
DP=35;MQ=250.00;MQRankSum=4.983;ReadPosRankSum=3.217;FractionInf
ormativeReads=1.000;R2_5P_bias=0.000
GT:AD:AF:DP:F1R2:F2R1:GQ:PL:SPL:ICNT:GP:PRI:SB:MB:PS
0|1:20,15,0:0.429:35:9,7,0:11,8,0:47:83,0,50,572,758,622:255,0,2
55:19,0:4.844e+01,8.387e-
05,5.300e+01,4.500e+02,4.500e+02,4.500e+02:0.00,34.77,37.77,34.7
7,69.54,37.77:11,9,10,5:12,8,8,7:1947645
chr1 1947648 . G A,<NON_REF> 50.00 PASS
DP=36;MQ=250.00;MQRankSum=5.078;ReadPosRankSum=2.563;FractionInf
ormativeReads=1.000;R2_5P_bias=0.000
GT:AD:AF:DP:F1R2:F2R1:GQ:PL:SPL:ICNT:GP:PRI:SB:MB:PS
1|0:16,20,0:0.556:36:8,9,0:8,11,0:48:85,0,49,734,613,698:255,0,2
55:16,0:5.000e+01,7.067e-
05,5.204e+01,4.500e+02,4.500e+02,4.500e+02:0.00,34.77,37.77,34.7
7,69.54,37.77:10,6,11,9:8,8,12,8:1947645
During the genotyping step, all haplotypes and all variants are considered over an active region. For
each pair of variants, if both variants occur on all of the same haplotypes, or if either is a homozygous
variant, then they are phased together. If the variants only occur on different haplotypes, then they are
phased opposite to each other. If any heterozygous variants are present on some of the same
haplotypes but not others, phasing is aborted and no phasing information is output for the active
region.
Ploidy Support
The small variant caller currently only supports either ploidy 1 or 2 on all contigs within the reference
except for the mitochondrial contig, which uses a continuous allele frequency approach (see
Mitochondrial Calling on page 88). The selection of ploidy 1 or 2 for all other contigs is determined as
follows.
• If --sample-sex is not specified on the command line, the Ploidy Estimator determines the sex. If
the Ploidy Estimator cannot determine the sex karyotype or detects sex chromosome aneuploidy,
all contigs are processed with ploidy 2.
• If --sample-sex is specified on the command line, contigs are processed as follows.
– For female samples, DRAGEN processes all contigs with ploidy 2, and marks variant calls on
chrY with a filter PloidyConflict.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
87
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

– For male samples, DRAGEN processes all contigs with ploidy 2, except for the sex
chromosomes. DRAGEN processes chrX with ploidy 1, except in the PAR regions, where it is
processed with ploidy 2. chrY is processed with ploidy 1 throughout.
DRAGEN detects sex chromosomes by the naming convention, either X/Y or chrX/chrY. No other
naming convention is supported.
--sample-sex Ploidy Estimation Sample Sex in Small VC
male Not relevant Male
female Not relevant Female
Not specified XY Male
Not specified XX Female
Not specified Everything else None
Overlapping Mates in Small Variant Calling
DRAGEN v3.6 and later no longer treats overlapping mates as independent evidence for a given event.
This improves how overlapping mates in a read pair are handled in the variant caller and accuracy.
DRAGEN handles overlapping mates as follows in both the germline and somatic pipelines.
• When the two overlapping mates agree with each other on the allele with the highest HMM score,
the genotyper uses the mate with the greatest difference between the highest and the second
highest HMM score. The HMM score of the other mate becomes zero.
• When the two overlapping mates disagree, the genotyper sums the HMM score between the two
mates, assigns the combined score to the mate that agrees with the combined result, and changes
the HMM score of the other mate to zero.
• The base qualities of overlapping mates are no longer adjusted.
Mitochondrial Calling
Typically, there are approximately 100 mitochondria in each mammalian cell, and each mitochondrion
harbors 2–10 copies of mitochondrial DNA (mtDNA). For example, if 20% of the chrM copies have a
variant, then the allele frequency (AF) is 20%. This is also referred to as continuous allele frequency.
The expectation is that the AF of variants on chrM is anywhere between 0% and 100%.
DRAGEN now processes chrM through a continuous AF pipeline. In this case, a single ALT allele is
considered, and the AF is estimated, and can be anywhere between 0% and 100%.
QUAL is not output in the chrM variant records. Instead, the confidence score is INFO/LOD
##INFO=<ID=LOD,Number=1,Type=Float,Description="Variant LOD score">
which gives the confidence that a variant is present at a given locus.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
88
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
GQ is not output in the chrM variant records because DRAGEN doesn't test for multiple diploid genotype
candidates. Instead, an ALT allele is considered as a candidate variant, and if INFO/LOD > vc-lod-call-
threshold (default = 4), then the FORMAT/GT is hard-coded to 0/1, and the FORMAT/AF yields an
estimate on the variant allele frequency, which ranges anywhere within [0,1].
The following filters can be applied to mitochondrial variant calls.
• --vc-lod-call-threshold
LOD threshold for emitting calls in the VCF. The default is 4.
• --vc-lod-filter-threshold
LOD threshold to mark emitted VCF calls as filtered. The default is 6.3.
• If INFO/LOD < vc-lod-call-threshold, the variant is not emitted in the VCF.
• If INFO/LOD > vc-lod-call-threshold but INFO/LOD < vc-lod-filter-threshold the variant is emitted in
the VCF but FILTER=lod_fstar.
• If INFO/LOD > vc-lod-call-threshold and INFO/LOD > vc-lod-filter-threshold, the variant is emitted
in the VCF and FILTER=PASS.
The following are example VCF records on the chrM, showing one call with very high AF and another
with very low AF. In both cases FORMAT/LOD > emit_threshold. FORMAT/LOD is also > lod_fstar
threshold, so the FILTER annotation is PASS.
chrM 2259 . C T . PASS
DP=9791;MQ=60.00;LOD=38838.40;FractionInformativeReads=0.994
GT:AD:AF:F1R2:F2R1:DP:SB:MB
0/1:5,9729:0.999:1,5007:4,4722:9734:3,2,4885,4844:1,4,4807,4922
chrM 16192 . C T . PASS
DP=9644;MQ=60.00;LOD=26.12;FractionInformativeReads=0.992
GT:AD:AF:F1R2:F2R1:DP:SB:MB
0/1:9537,26:0.003:5530,16:4007,10:9563:4484,5053,13,13:4961,4576,19,
gVCF and Joint VCF Mode
In gVCF mode, which is available for the germline pipeline, the NON_REF regions are output alongside
the variant records. The following is an example of NON_REF and variant regions output in chrM gVCF
mode:
chrM 751 . A <NON_REF> . PASS END=1437 GT:AD:DP:GQ:MIN_DP:PL:SPL:ICNT
0/0:6920,9:6929:99:4077:0,120,1800:0,255,255:40,4
chrM 1438 . A G,<NON_REF> . PASS
DP=8500;MQ=57.39;LOD=30441.87;FractionInformativeReads=0.871
GT:AD:AF:F1R2:F2R1:DP:SB:MB
0/1:0,7400,0:1.000:0,3765,0:0,3635,0:7400:0,0,3994,3406:0,0,3633,3767
chrM 1439 . A <NON_REF> . PASS END=2258 GT:AD:DP:GQ:MIN_DP:PL:SPL:ICNT
0/0:6120,10:6130:99:4190:0,120,1800:0,255,255:40,14
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
89
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
FORMAT/GT
In NON_REF regions, the FORMAT/GT is hard-coded to 0/0 and at variant loci, the FORMAT/GT is hard-
coded to 0/1.
The FORMAT/GT for chrM is not determined by FORMAT/AF. It is determined by whether a variant is
emitted at a position or not. The decision to emit the variant or not is determined by comparing the
INFO/LOD score to a threshold. All variants with FORMAT/LOD > emit_threshold get emitted. Variants
with FORMAT/LOD < emit_threshold are not emitted in the gVCF and get banded in a NON_REF region
with FORMAT/GT=0/0.
One of the following two scenarios can occur at a given position.
• A variant is detected by the genotyper at a given position, and the FORMAT/LOD > emit_threshold.
In this case, the FORMAT/GT is hard-coded to 0/1, and DRAGEN outputs FORMAT/AD, FORMAT/DP
and FORMAT/AF computed at that position.
• No variant is detected at the given position, or, a variant is detected but the FORMAT/LOD < emit_
threshold. In this case, the FORMAT/GT is hard-coded to 0/0, and the position gets banded with
contiguous positions if they are in the same scenario. All positions with FORMAT/GT = 0/0 get
banded together. The FORMAT/DP reported for the band is computed as the median DP across all
positions within the band. The FORMAT/AD values reported in the band are the AD values selected
at the position where FORMAT/DP = median DP. In the joint VCF, FORMAT/AF is computed based
on FORMAT/AD.
The following are examples of variant record on chrM in a trio joint VCF.
The first sample shows a variant, but FORMAT/FT is lod_fstar because FORMAT/LOD < lod_fstar_
threshold.
The second sample does not have a variant and the FORMAT/AF=0.
The third sample does not have a variant even though the FORMAT/AF>0, this means that that position
for the sample is in a NON_REF region over which no variant was detected with sufficient confidence.
chrM 2622 . G A . . DP=11199;MQ=59.73 GT:AD:AF:DP:FT:LOD:F1R2:F2R1
0/1:3375,8:0.002:3383:lod_fstar:4.4:1689,2:1686,6
0/0:5629,1:0:5118:PASS:.:.:. 0/0:3505,8:0.003:2645:PASS:.:.:.
QUAL, QD, and GQ Formulation
In single sample VCF and gVCF, the QUAL follows the definition of the VCF specification
(https://samtools.github.io/hts-specs/VCFv4.3.pdf).
• QUAL is the Phred-scaled probability that the site has no variant and is computed as:
QUAL = -10*log10 (posterior genotype probability of a homozygous-
reference genotype (GT=0/0))
That is, QUAL = GP (GT=0/0), where GP = posterior genotype probability in Phred scale.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
90
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

QUAL = 20 means there is 99% probability that there is a variant at the site. The GP values are also
given in Phred-scale in the VCF file.
• GQ is the Phred-scaled Probability that the call is incorrect.
GQ=-10*log10(p), where p is the probability that the call is incorrect.
GQ=-10*log10(sum(10.^(-GP(i)/10))) where the sum is taken over the GT that did not win.
So, GQ of 3 means there's a 50 percent chance that the call is incorrect, and GQ of 20 means
there's a 1 percent chance that the call is incorrect.
• QD is the QUAL normalized by the read depth, DP.
Metric QUAL GQ QD
Description Probability that the site has no
variant
Probability that the call is
incorrect
Qual
normalized by
Depth
Formulation QUAL = GP(GT=0/0) GQ=-10*log10(p) QUAL/DP
Scale Unsigned Phred Unsigned Phred Unsigned
Phred
Numerical
example
QUAL=20: 1 % chance that there is
no variant at the site
QUAL=50: 1 in 1e5 chance that
there is no variant at the site
GQ=3, 50% chance that
the call is incorrect
GQ=20, 1% chance that
the call is incorrect
Change in the Range of Values Between DRAGEN 2.6+ and Earlier Versions of
DRAGEN/GATK
The range of the QUAL (and therefore GQ and QD) values has changed between older DRAGEN
versions (and/or GATK) and DRAGEN 2.6 and later versions. In older DRAGEN versions, the QUAL
calculations followed the GATK approach. In newer DRAGEN versions (2.6 and later), the algorithms for
small variant detection were improved and they result in more realistic QUAL (and therefore GQ and
QD) values (ie, smaller values than GATK).
The QUAL values changed because the probability calculations used by GATK and earlier DRAGEN
versions assume that errors are uncorrelated across reads. This is assumed for both mapping errors
and base-call errors. In a real world with correlated errors, this leads to wildly inflated QUAL scores as
well as suboptimal detection performance. The algorithms in DRAGEN 2.6 and later incorporate the
hypothesis of correlated errors from within the variant caller, leading to more accurate detection and
more realistic QUAL scores. The more realistic QUAL scores in DRAGEN 2.6+ are smaller than the
inflated QUAL scores in GATK.
Histogram of QUAL, QD, and GQ
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
91
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
When plotting the histogram of QUAL (and QD and GQ) values across the calls of a DRAGEN 2.6+ VCF,
a peak can occur at a certain value (eg, 50) with a smaller portion of QUAL values below and above this
level. This is because the Foreign Read Detection (FRD) algorithm sets a limit on the QUAL (and
indirectly on GQ and QD) value of heterozygous variants. See Foreign Read Detection on page 92. The
exact value depends on the mapping between the maximum MAPQ from the mapper and a phred
confidence that a read is correctly mapped. Homozygous variants are not subject to this limit, because
the FRD hypothesis is applied against heterozygous variants only (this in turn is because the --vc-frd-
beta-max option that specifies the maximum allele frequency for the foreign read hypothesis is set to
0.5).
The high-level summary is that the QUAL of heterozygous variants is bounded, and the QUAL of
homozygous variants is not. However, the QUAL scale is the same across all variants (het or hom), only
the bound is different.
Modeling of Correlated Errors Across Reads
DRAGEN has two algorithms that model correlated errors across reads in a given pileup.
• Foreign read detection (FRD) detects mismapped reads. FRD modifies the probability calculation to
account for the possibility that a subset of the reads were mismapped. Instead of assuming that
mapping errors occur independently per read, it estimates the probability that a burst of reads is
mismapped, incorporating such evidence as MAPQ and skewed AF. For more information, see
Foreign Read Detection on page 92.
• The base quality drop off (BQD) algorithm detects correlated base call errors using a mechanism
that detects systematic and correlated base call errors caused by the sequencing instrument. The
detection mechanism exploits certain properties of those errors (strand bias, position of the error in
the read, base quality) to estimate the probability that the alleles are the result of a systematic
error event rather than a true variant. For more information, see Base Quality Dropoff on page 93.
Foreign Read Detection
Mapping errors typically occur in bursts, but variant callers typically treat mapping errors as
independent error events per read, which can result in high confidence scores in spite of low MAPQ
and/or skewed AF. To mitigate overestimation of confidence scores some variant callers include a
threshold on the minimum MAPQ to include in the calculation, however this method can discard
evidence and result in false positives.
DRAGEN includes Foreign Read Detection (FRD), which is an extends the legacy genotyping algorithm
by incorporating an additional hypothesis that reads in the pileup might be foreign reads (ie, their true
location is elsewhere in the reference genome). The algorithm exploits multiple properties (skewed
allele frequency and low MAPQ) and incorporates this evidence into the probability calculation.
Sensitivity is improved by rescuing FN, correcting genotypes, and enabling lowering of the MAPQ
threshold for incoming reads into the variant caller. Specificity is improved by removing FP and
correcting genotypes.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
92
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Base Quality Dropoff
Conventional variant callers treat sequencing errors as independent across reads, however bursts of
errors can occur at a specific locus and increase the number of false positives.
These errors have distinct characteristics differentiating them from true variants. The base quality drop
off (BQD) algorithm is a detection mechanism that exploits certain properties of those errors (strand
bias, position of the error in the read, low mean base quality over said subset of reads at the locus of
interest) and incorporates them into the probability calculation.
Joint Detection of Overlapping Variants
Genotyping can benefit if variants at multiple loci in a single active region are detecting jointly. Joint
Detection feature groups loci in a Joint Detection region if the following conditions are met:
• Loci have alleles that overlap each other.
• Loci are in the STR region or less than 10 bases apart of the STR region.
• Loci are less than 10 bases apart of each other.
Joint Detection alters the variant caller algorithm in the part of localized haplotype assembly and
genotyping. It generates a haplotype list where all possible combinations of the alleles in the Joint
Detection regions are represented. This leads to larger number of haplotypes. During genotyping, Joint
Detection calculates likelihoods that each haplotype pair is the truth, given the read pileup is observed.
Genotypes likelihoods are calculated as sum of the likelihoods of haplotype pairs that support the
alleles in the genotypes. Genotypes with maximum likelihood are reported.
You can enable Joint Detection by setting the --vc-enable-joint-detection option to true. Run time
slightly increases when Joint Detection feature is enabled.
ROHCaller
Regions of homozygosity (ROH) are detected as part of the small variant caller. The caller detects and
outputs the runs of homozygosity from whole genome calls on autosomal human chromosomes. Sex
chromosomes are ignored. ROH output allows downstream tools to screen for and predict
consanguinity between the parents of the proband subject.
A region is defined as consecutive variant calls on the chromosome with no large gap in between these
variants. In other words, regions are broken by chromosome or by large gaps with no SNV calls. The
gap size is set to 3 Mbases.
ROH Algorithm
The ROH algorithm runs on the small variant calls. It excludes variants with multiallelic sites, indels,
complex variants, non-PASS filtered calls, and homozygous reference sites. The variant calls are then
filtered further using a blacklist bed, and finally depth filtering is applied after the blacklist filter. The
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
93
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
default value for the fraction of filtered calls is 0.2, filtering the calls with the highest 10% and lowest
10% in DP values. The resulting calls are then used to find regions.
The ROH algorithm first finds seed regions that contain at least 50 consecutive homozygous SNV calls
with no heterozygous SNV or gaps of 500,000 bases between the variants. The regions can be
extended using a scoring system that functions as follows.
• Score increases with every additional homozygous variant (0.025) and decreases with a large
penalty (1–0.025) for every heterozygous SNV. This provides some tolerance of presence of
heterozygous SNV in the region.
• Each region expands on both ends until the regions reach the end of a chromosome, a gap of
500,000 bases between SNVs occurs, or the score becomes too low (0).
Overlapping regions are merged into a single region. Regions can be merged across gaps of 500,000
bases between SNVs if a single region would have been called from the beginning of the first region to
the end of the second region without the gap. There is no maximum size for regions, but regions
always end at chromosome boundaries.
ROH Options
• --vc-enable-roh
Enable or disable the ROHcaller by setting this option to true or false. Enabled by default for human
autosomes only.
• --vc-roh-blacklist-bed
If provided, the ROH caller ignores variants that are contained in any region in the blacklist BED file.
DRAGEN distributes blacklist files for all popular human genomes and automatically selects a
blacklist to match the genome in use, unless this option is used explicitly select a file.
ROH Output
The ROH caller produces an ROH output file named <output-file-prefix>.roh.bed in which each
row represents one region of homozygosity. The bed file contains the following columns:
Chromosome Start End Score #Homozygous #Heterozygous
Where
• Score is a function of the number of homozygous and heterozygous variants, where each
homozygous variant increases the score by 0.025, and each heterozygous variant reduces the
score by 0.975.
• Start and end positions are a 0-based, half-open interval.
• #Homozygous is number of homozygous variants in the region.
• #Heterozygous is number of heterozygous variants in the region.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
94
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The caller also produces a metrics file named <output-file-prefix>.roh_metrics.csv that lists
the number of large ROH and percentage of SNPs in large ROH (> 3 MB).
B-Allele Frequency Output
B-Allele frequency (BAF) output is enabled by default in germline and somatic VCF and gVCF runs.
The BAF value is equal to either AF or (1 – AF), where
• AF = (alt_count / (ref_count + alt_count))
• BAF = 1 – AF, only when ref base < alt base, order of priority for bases is A < T <G < C < N
For each small variant VCF entry with exactly one SNP alternate allele, the output contains a
corresponding entry in the BAF output file.
• <NON_REF> lines are excluded
– ForceGT variants (as marked by the "FGT" tag in the INFO field) are not included in the output,
unless the variant also contains the "NML" tag in the INFO field.
– Variants where the ref_count and alt_count are both zero are not included in the output.
BAF Options
--vc-enable-baf
Enable or disable B-allele frequency output. Enabled by default.
BAFOutput
The BF generates are BigWig-compressed files, named <output-file-prefix>.baf.bw and
<output-file-prefix>.hard-filtered.baf.bw. The hard-filtered file only contains entries for
variants that pass the filters defined in the VCF (ie, PASS entries).
Each entry contains the following information:
Chromosome Start End BAF
Where:
• Chromosome is a string matching a reference contig.
• Start and end values are zero-based, half open intervals.
• BAF is a floating point value.
Somatic mode
The DRAGEN Somatic Pipeline allows ultrarapid analysis of NGS data to identify cancer-associated
mutations in somatic chromosomes. DRAGEN calls SNVs and indels from both matched tumor-normal
pairs and tumor-only samples.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
95
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
For the tumor-normal pipeline, both samples are analyzed jointly such that germline variants are
excluded, generating an output specific to tumor mutations. The tumor-only pipeline produces a VCF
file containing both germline and somatic variants that can be further analyzed to identify tumor
mutations. For the tumor sample, both pipelines make no ploidy assumptions, enabling detection of
low-frequency alleles.
The output, after multiple filtering dbSNP Annotation on page 115steps, is in the form of a VCF file.
Variants that fail the filtering steps are kept in the output VCF, with a FILTER annotation indicating
which filtering steps have failed.
You can use somatic quality (SQ) as the primary metric to describe the confidence with which the caller
made a somatic call. SQ is reported as a format field for the tumor sample. Variants with SQ score
below the SQ filter threshold are filtered out using the weak_evidence tag. To trade off sensitivity
against specificity, adjust the SQ filter threshold. Lower thresholds produce a more sensitive caller and
higher thresholds produce a more conservative caller.
Somatic Mode Options
Somatic mode has the following command line options:
• --tumor-fastq1 and --tumor-fastq2
The --tumor-fastq1 and --tumor-fastq2 options are used to input a pair of FASTQ files into the
mapper aligner and somatic variant caller. These options can be used with OTHER FASTQ options
to run in tumor-normal mode. For example:
dragen -f -r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--tumor-fastq1 <TUMOR_FASTQ1> \
--tumor-fastq2 <TUMOR_FASTQ2> \
--RGID-tumor <RG0-tumor> -–RGSM-tumor <SM0-tumor> \
-1 <NORMAL_FASTQ1> \
-2 <NORMAL_FASTQ2> \
--RGID <RG0> –RGSM <SM0> \
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M
• --tumor-fastq-list
The --tumor-fastq-list option is used to input a list of FASTQfiles into the mapper aligner and
somatic variant caller.This option can be used with other FASTQoptions to run in tumor-normal
mode. For example:
dragen -f \
-r /staging/human/reference/hg19/hg19.fa.k_21.f_16.m_149 \
--tumor-fastq-list <TUMOR_FASTQ_LIST> \
--fastq-list <NORMAL_FASTQ_LIST> \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
96
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--enable-variant-caller true \
--output-directory /staging/examples/ \
--output-file-prefix SRA056922_30x_e10_50M
• --tumor-bam-input and --tumor-cram-input
The --tumor-bam-input or --tumor-cram-input option is used to input a mapped BAM or CRAM file
into the somatic variant caller.These options can be used with other BAM/CRAM options to run in
tumor-normal mode.
• --vc-min-tumor-read-qual
The --vc-min-tumor-read-qual option specifies the minimum read quality (MAPQ) to be considered
for variant calling. The default value is 3 for tumor-normal analysis or 20 for tumor-only analysis.
• --vc-callability-tumor-thresh and --vc-callability-normal-thresh
The --vc-callability-tumor-thresh option specifies the callability threshold for tumor samples, and
the --vc-callability-normal-thresh option specifies the callability threshold for normal samples, if
present. The somatic callable regions report includes all regions with tumor coverage above the
tumor threshold and if applicable, normal coverage above the normal threshold. For more
information on the somatic callable regions report, see Somatic Callable Regions Report on page
203. The default value for the tumor-only threshold is 15 and the tumor-normal threshold is 5.
• --vc-somatic-hotspots and --vc-hotspot-log10-prior-boost
The somatic hotspots option allows an input VCF to specify the positions where the risk for somatic
mutations are assumed to be significantly elevated. DRAGEN genotyping priors are boosted for all
postions specified in the VCF, so it is possible to call a variant at one of these sites with fewer
supporting reads. The cosmic database in VCF format can be used as one source of prior
information to boost sensitivity for known somatic mutations. The size of the adjustment can be
controlled via vc-hotspot-log10-prior-boost, which has a default value of 4 (log10 scale)
corresponding to an increase of 40 phred.
• --vc-enable-liquid-tumor-mode and vc-tin-contam-tolerance
In a tumor-normal analysis, DRAGEN accounts for tumor-in-normal (TiN) contamination by running
liquid tumor mode. Liquid tumor mode is disabled by default. When liquid tumor mode is enabled,
DRAGEN is able to call variants in the presence of TiN contamination up to a specified maximum
tolerance level. --vc-enable-liquid-tumor-mode enables liquid tumor mode with a default
maximum contamination TiN tolerance of 0.15. If using the default maximum contamination TiN
tolerance, somatic variants are expected to be observed in the normal sample with allele
frequencies up to 15% of the corresponding allele in the tumor sample. vc-tin-contam-
tolerance enables liquid tumor mode and allows you to set the maximum contamination TiN
tolerance. The maximum contamination TiN tolerance must be greater than zero. For example, vc-
tin-contam-tolerance=-0.1.
Liquid tumors in liquid tumor mode refers to hematological cancers, such as leukemia. For liquid
tumors it is not feasible to use blood as a normal control because the tumor is present in the blood.
Skin or saliva is typically used as the normal sample. However, skin and saliva samples can still
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
97
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
contain blood cells, so that the matched normal control sample contains some traces of the tumor
sample and somatic variants are observed at low frequencies in the normal sample. If the
contamination is not accounted for, it can severely impact sensitivity by suppressing true somatic
variants.
In typical use cases of liquid tumor mode, the library is WGS or WES, with medium depth for
example (100x T/ 40xN), and the lowest VAF detected for these types of depths is ~5%. In typical
use cases of liquid biopsy, the library is a targeted gene panel (eg 500 genes), with very high raw
depth (> 2000-5000x), and uses UMI indexing to enable sensitivity at VAF down to 0.5 % LoD.
Unique Molecular Identifiers Support
To optimize variant calling for different UMI use cases, use the following two batch modes. Both
options are false by default. Enable UMI variant calling by setting one of them to true.
• --vc-enable-umi-solid—The VC UMI solid mode is optimized for solid tumors with post collapsed
coverage rates of ~200—300X and target allele frequencies of 5% and higher.
• --vc-enable-umi-liquid—The liquid biopsy pipeline is not equivalent to liquid tumor mode. The liquid
biopsy pipeline starts from a regular blood sample and looks for low VAF somatic variants from
tumor cell free DNA floating in the blood. This type of test enables tumor profiling
(diagnosis/biomarker identification) from plasma rather than from tissue, which requires an
invasive biopsy. The VC UMI liquid mode is optimized for a liquid biopsy pipeline with post collapsed
coverage rates of ~2000–2500X and target allele frequencies of 0.4% and higher.
These batch settings do not include the vc-systematic-noise filter. DRAGEN recommends adding the
filter. For more information, see Systematic Noise Filtering on page 115.
In UMI pipelines, DRAGEN can estimate nucleotide mutation biases, such as oxidation and deamination
artifacts, that might exist upstream of the sequencing system on a per sample basis. During variant
calling, DRAGEN corrects the biases.
To enable this feature, use --vc-target-bed, which also specifies the target regions for variant calling,
or --vc-snp-error-cal-bed. If using --vc-snp-error-cal-bed, specify the regions to estimate nucleotide
substitution bias from if different from the target bed file or if a target bed file is not specified. If a third-
party tool is used to produce the collapsed reads, then configure the tool so that the base call quality
scores quantify the error shown on the sequencing system only. DRAGEN uses this error estimation to
account for errors upstream of the sequencing system.
Post Somatic Calling Filtering
The following options are available for post somatic calling filtering:
• --vc-sq-call-threshold
Emits calls in the VCF. The default is 3.0 for tumor-normal and 0.1 for tumor-only. If the value for
vc-sq-filter-threshold is lower than vc-sq-call-threshold, the filter threshold value is used instead of
the call threshold value.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
98
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• --vc-sq-filter-threshold
Marks emitted VCF calls as filtered. The default is 17.5 for tumor-normal and 6.5 for tumor-only.
• --vc-enable-triallelic-filter
Enables the multiallelic filter.The default is true.
• --vc-enable-af-filter
Enables the allele frequency filter. The default value is false. When set to true, the VCF excludes
variants with allele frequencies below the AF call threshold or variants with an allele frequency
below the AF filter threshold and tagged with low AF filter tag. The default AF call threshold is 1%
and the default AF filter threshold is 5%. To change the threshold values, use the following
command line options: vc-af-call-threshold and vc-af-filter-threshold.
• --vc-enable-non-homref-normal-filter
Enables the non-homref normal filter. The default value is true. When set to true, the VCF filters out
variants if the normal sample genotype is not a homozygous reference.
• --vc-enable-vaf-ratio-filter
Adds one condition to be filtered out by the alt_allele_in_normal filter. The default value is false.
When set to true, the VCF filters out variants if the normal sample AF is greater than 20% of tumor
sample AF.
Somatic
Mode
Filter ID Description
Tumor-
Only &
Tumor-
Normal
clustered_events Clustered events were observed in a given active
region. Threshold for clustered events is configurable
(default is ≥ 3). Enabled only when using --vc-enable-
gatk-acceleration=true.
Tumor-
Only &
Tumor-
Normal
weak_evidence Variant does not meet likelihood threshold. The
likelihood ratio for SQ tumor-normal is < 17.5 or < 3.0
for SQ tumor-only.
Tumor-
Only &
Tumor-
Normal
multiallelic Site filtered if there are two or more alt alleles at this
location in the tumor.
Tumor-
Only &
Tumor-
Normal
str_contraction Suspected PCR error where the alt allele is one repeat
unit less than the reference. Enabled only when using -
-vc-enable-gatk-acceleration=true.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
99
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Somatic
Mode
Filter ID Description
Tumor-
Only &
Tumor-
Normal
base_quality Median base quality of alt reads at this locus is < 20.
Tumor-
Only &
Tumor-
Normal
mapping_quality Median mapping quality of alt reads at this locus is < 20
(tumor-normal) or < 30 (tumor-only).
Tumor-
Only &
Tumor-
Normal
fragment_length Absolute difference between the median fragment
length of alt reads and median fragment length of ref
reads at a given locus > 10000.
Tumor-
Only &
Tumor-
Normal
read_position Median of distances between the start and end of read
and a given locus < 5 (the variant is too close to edge of
all the reads).
Tumor-
Only &
Tumor-
Normal
panel_of_normals Seen in at least one sample in the panel of normal VCF.
Tumor-
Only &
Tumor-
Normal
low_af Allele frequency is below the threshold specified with --
vc-af-filter-threshold (default is 5%). Enabled only
when using --vc-enable-af-filter=true.
Tumor-
Only &
Tumor-
Normal
systematc_noise If AQ score is < 10 (default) for tumor-normal or < 60
(default) for tumor-only, the site is filtered.
Matched Normal Control Sample Post Somatic Filtering
Somatic
Mode
Filter ID Description
Tumor-
Normal
noisy_normal More than three alleles are observed in the normal
sample at allele frequency above 9.9%.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
100
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Somatic
Mode
Filter ID Description
Tumor-
Normal
alt_allele_in_normal Alt allele frequency in the normal sample is above
0.2 plus the maximum contamination tolerance. For
solid tumor mode the value is 0. For liquid tumor
mode the default value is 0.15. See vc-enable-vaf-
ratio-filter for optional conditions.
Tumor-
Normal
filtered_reads More than 90% of reads have been filtered out.
Tumor-
Normal
no_reliable_supporting_
read
No reliable supporting read was found in the tumor
sample. A reliable supporting read is a read
supporting the alt allele with mapping quality ≥ 40,
fragment length ≤ 10,000, basecall quality ≥ 25, and
distance from start/end of read ≥ 5.
Tumor-
Normal
strand_artifact Severe strand bias. Alt allele supported by at least
four reads, but only on one strand.
Tumor-
Normal
too_few_supporting_reads Variant is supported by < 3 reads in the tumor
sample.
Tumor-
Normal
non-homref-normal Normal sample genotype is not a homozygous
reference.
Tumor-
Normal
germline_risk Likelihood of allele being present in normal > 0.025.
Enabled only when using --vc-enable-gatk-
acceleration=true.
Tumor-
Normal
artifact_in_normal TLOD of the normal read set (Normal artifact LOD)
is > 0.0. Not called normal artifact if allele fraction in
normal is much smaller than allele fraction in tumor
(normalAlleleFraction < (0.1 * tumorAlleleFraction).
Enabled only when using --vc-enable-gatk-
acceleration=true.
QUAL is not output in the somatic variant records. Instead, the confidence score is FORMAT/SQ.
##FORMAT=<ID=SQ,Number=1,Type=Float,Description="Somatic quality">
The field is specific to the sample. For the tumor samples, it quantifies the evidence that a somatic
variant is present at a given locus.
If a normal sample is also available, the corresponding FORMAT/SQ value quantifies the evidence that
the normal sample is a homozygous reference at a given locus.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
101
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
GQ is not output in the somatic variant records, because DRAGEN does not test for multiple diploid
genotype candidates. Instead, an ALT allele is considered as a candidate somatic variant. If tumor SQ >
vc-sq-call-threshold (default is 3), then the FORMAT/GT for the tumor sample is hard-coded to 0/1, and
the FORMAT/AF yields an estimate on the somatic variant allele frequency, which ranges anywhere
within [0,1].
• If tumor SQ < vc-sq-call-threshold, the variant is not emitted in the VCF.
• If tumor SQ > vc-sq-call-threshold but tumor SQ < vc-sq-filter-threshold, the variant is emitted in
the VCF, but FILTER=weak_evidence.
• If tumor SQ > vc-sq-call-threshold and tumor SQ > vc-sq-filter-threshold, the variant is emitted in
the VCF and FILTER=PASS (unless the variant is filtered by a different filter).
• The default vc-sq-filter-threshold is 17.5 for tumor-normal and 3.0 for tumor-only analysis.
The following is an example somatic T/N VCF record. Tumor SQ > vc-sq-call-threshold but tumor SQ <
vc-sq-filter-threshold, so the FILTER is marked as weak_evidence.
2 593701 . G A . weak_evidence
DP=97;MQ=48.74;SQ=3.86;NLOD=9.83;FractionInformativeReads=1.000
GT:SQ:AF:F1R2:F2R1:DP:SB:MB 0/0:9.83:33,0:0.000:14,0:19,0:33
0/1:3.86:61,3:0.047:29,2:32,1:64:35,26,0,3:39,22,1,2
The clustered-events penalty is an exception to the above rule for emitting variants. By default, the
clustered-events penalty replaces the clustered-events filter in tumor-normal mode. Instead of
applying a hard filter when too many events are clustered together, DRAGEN applies a penalty to the
SQ scores of co-phased clustered events. Clustered events with weak evidence are no longer called,
but clustered events with strong evidence can still be called. This is equivalent to lowering the prior
probability of observing clustered co-phased variants. The penalty is applied after the decision to emit
variants, so that penalized variants still appear in the VCF if their unpenalized score is high enough.
Joint Analysis for Multiple Samples
DRAGEN supports pedigree-based and population-based joint analysis for multiple samples. In
pedigree-based analysis, samples from the same species are related to each other. In population-
based analysis, samples of the same species are unrelated to each other.
Joint analysis requires a gVCF file for each sample. To create a gVCF file, run the germline small variant
caller with the --vc-emit-ref-confidence gVCF option.
The gVCF file contains information on the variant positions and positions determined to be
homozygous to the reference genome. For homozygous regions, the gVCF file includes statistics that
indicate how well reads support the absence of variants or alternative alleles. Contiguous homozygous
runs of bases with similar levels of confidence are grouped into blocks, referred to as hom-ref blocks.
Not all entries in the gVCF are contiguous. A reference might contain gaps that are not covered by
either variant line or a hom-ref block. Gaps correspond to regions that are not callable. A region is not
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
102
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

callable if there is not at least one read mapped to the region with a MAPQ score above zero. The
following example shows a joint VCF where one sample has a variant, and the other two samples are in
a gVCF gap. Gaps are represented by "./.:.:".
1 605262 . G A 13.41 DRAGENHardQUAL
AC=2;AF=1.000;AN=2;DP=2;FS=0.000;MQ=14.00;QD=6.70;SOR=0.693
GT:AD:AF:DP:GQ:FT:F1R2:F2R1:PL:GP ./.:.:.:.:.:LowDepth
1/1:0,2:1.000:2:4:PASS:0,0:0,2:50,6,0:1.383e+01,4.943e+00,1.951e+00
./.:.:.:.:.:LowDepth
Pedigree Mode
Use pedigree mode to jointly analyze samples from related individuals and to perform de novo calling.
To invoke pedigree mode set the --enable-joint-genotyping option to true and use the --pedigree-file
option to specify the path to a pedigree file that describes the relationship between panels.
The pedigree file must be a tab-delimited text file with the file name ending in the .ped extension. The
following information is required.
Column Header Description
Family_ID The pedigree identifier.
Individual_ID The ID of the individual.
Paternal_ID The ID of the individual's father. If the founder, the value is 0.
Maternal_ID The ID of the individual's mother. If the founder, the value is 0.
Sex The sex of the sample. If male, the value is 1. If female, the value
is 2.
Phenotype The genetic data of the sample. If unknown, the value is 0. If
unaffected, the value is 1. If affected, the value is 2.
The following is an example of an input pedigree file.
#Family_ID Individual_ID Paternal_ID Maternal_ID Sex
Phenotype
FAM001 NA12877_Father 0 0 1 1
FAM001 NA12878_Mother 0 0 2 1
FAM001 NA12882_Proband NA12877_Father NA12878_Mother 2 2
FAM001 NA12883_Proband NA12877_Father NA12878_Mother 1 0
To compare multiple pedigrees, you can run gVCF Genotyper on the output of Joint Genotyper and
merge multiple joint-called pedigrees into a single multisample VCF.
Use the --vc-emit-ref-confidence gVCF option to configure the Joint Genotyper to write a
multisample GVCF.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
103
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
De Novo Calling
The De Novo Caller identifies all the trios within the pedigree and generate a de novo score for each
child. The De Novo Caller supports multiple trios within a single pedigree. Pedigree Mode supports de
novo calling for small, structural, and copy number variants.
Pedigree Mode is run in multiple steps. The following is an example workflow for a trio from FASTQ.
1. Run single sample alignment and variant calling to generate per sample output using the following
inputs for Pedigree Mode.
– gVCF files for Small Variant Caller.
– *.tn.tsv files for the Copy Number Caller.
– BAM files for the Structural Variant Caller.
2. Run Pedigree Mode for Small Variant Caller.
For more information, see Small Variant De Novo Calling on page 104.
3. Run Pedigree Mode for Copy Number Caller.
For more information, see Multisample CNV Calling on page 1.
4. Run Pedigree Mode for Structural Variant Caller.
For more information, see Structural Variant De Novo Quality Scoring on page 176.
5. Run DeNovo Variant Small Variant Filtering.
For more information, see De Novo Small Variant Filtering on page 111.
Small Variant De Novo Calling
The Small Variant De Novo callers considers a trio of samples at a time, where samples are related via
a pedigree file. The Small Variant De Novo Caller determines all positions that have a Mendelian
conflict based on the genotype from the individual sample gVCFs. Sex chromosomes in males are
treated as haploid apart from the PAR regions, which are treated as diploid.
Each of those positions is then processed through the Pedigree Caller to compute a joint posterior
probability matrix for the possible genotypes. The probabilities are used to determine whether the
proband has a de novo variant with a DQ confidence score. All three subjects are assumed to have an
independent error probability.
At positions where the original genotype from the gVCFs shows a double Mendelian conflict (eg,
0/0+0/0->1/1 or 1/1+1/1->0/0), the trio samples genotypes can be adjusted to the highest joint posterior
probability that has at least one Mendelian conflict.
The DQ formula is DQ = -10log10(1 - Pdenovo).
Pdenovo is the sum of all indexes in the joint posterior probability matrix with one of more Mendelian
conflicts.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
104
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
In the GT overwrite step, it is possible that the parents GT to get overwritten. In the case of multiple
trios, the parents GT is based on the last trio processed. The trios are processed in the order they are
listed in the pedigree file. DRAGEN currently does not add an annotation in the VCF in cases where the
GT was overwritten.
The multisample VCF file is annotated with FORMAT/DQ and FORMAT/DN fields to the output a VCF
file that represents a de novo Quality Score and an associated de novo call. The DN field in the VCF is
used to indicate the de novo status for each segment.
The following are the possible values:
• Inherited—The called trio genotype is consistent with Mendelian inheritance.
• LowDQ—The called trio genotype is inconsistent with Mendelian inheritance and DQ is less than
the de novo quality threshold.
• DeNovo—The called trio genotype is inconsistent with Mendelian inheritance and DQ is greater
than or equal to the de novo quality threshold.
The following is an example VCF line for a trio:
1 16355525. G A 34.46 PASS
AC=1;AF=0.167;AN=6;DP=45;FS=6.69;MQ=108.04;MQRankSum=0.156;QD=2.46;ReadPos
RankSum=0;SOR=0.01
GT:AD:AF:DP:GQ:FT:F1R2:F2R1:PL:GP:PP:DPL:DN:DQ
0/1:11,3:0.214:14:39:PASS:8,2:3,1:74,0,47:39.454,0.00053613,49.99:0,1,104:
74,0,47:DeNovo:0.6737
0/0:18,0:0:16:48:PASS:.:.:0,48,605:.:0,12,224:0,48,255:.:0/0:14,0:0:14:42:
PASS:.:.:0,42,490:.:0,5,223:0,42,255:.:.
De Novo Small Variant Options
The following command line options are available for de novo small variant calling.
• --enable-joint-genotyping—Run the joint genotyping caller.
• --variant—Specify the gVCF input to the workflow.
• --pedigree-file—Specify the pedigree file to enable De Novo Calling if there is a trio present.
• --qc-snp-denovo-quality-threshold—Specify the minimum DQ value for a SNP to be considered de
novo. The default value is 0.05.
• --qc-indel-denovo-quality-threshold—Specify the minimum DQ value for an indel to be considered
de novo. The default value is 0.02.
Population Mode
DRAGEN provides a population-based analysis option to jointly analyze samples from unrelated
individuals. To initiate population mode, use the following genotypers.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
105
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
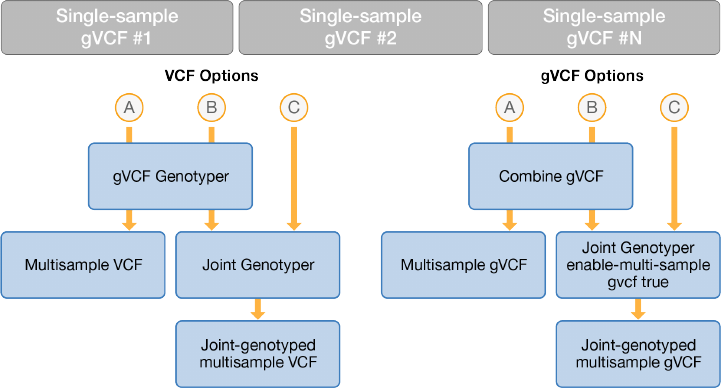
• gVCF Genotyper—Uses a set of single or multisample gVCFs as input and outputs a multisample
VCF, which contains one entry for any variant seen in any of the input gVCFs. The variants are
genotyped across all input samples using information from the hom-ref blocks as necessary. The
gVCF genotyper does not adjust genotypes based on population information. See gVCF Genotyper
Options on page 1 for information on the available command line options.
• Joint Genotyper—Uses information from the whole cohort to improve the accuracy of individual
genotypes. You can input multisample VCF, multisample gVCF, or a set of single sample gVCFs. To
receive output as a multisample gVCF, set --enable-multi-sample-gVCF to true. See Joint
Genotyper Options on page 1 for information on the available command line options.
• Combine gVCFs—Uses single-sample gVCF and multisample gVCFs to generate one multisample
gVCF. Variants present in any input sample are genotyped across all samples similarly to gVCF
genotyper. Combine gVCF accepts any combination of single-sample gVCFs and multisample
gVCFs as input, but generating output might be slow if merging a large number of samples. For
large scale population calling, use gVCF Genotyper instead of combine gVCF. See Combine gVCF
Options on page 1 for information on the available command line options.
The following figure displays the different pathways and data flows between the gVCF Genotyper,
Combine gVCFs, and Joint Genotyper.
To receive a list of variants present in the cohort and the genotypes of that variant in each of the cohort
members, run the gVCF Genotyper. Optionally, you can run the Joint Genotyper after to build a second
multisample VCF. The Joint Genotyper output refines the sample genotypes based on the population
information. If using the gVCF Genotyper output only, you can filter out infrequent variants to prevent
noise if the variants contain low depth or genotype quality. Use an open-source utility like bcftools on
the output file to filter the variants.
To compare multiple pedigrees, you can run gVCF Genotyper on the output of Joint Genotyper and
merge multiple joint-called pedigrees into a single multisample VCF.
Use the --vc-emit-ref-confidence gVCF option to configure the Joint Genotyper to write a
multisample GVCF.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
106
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Process gVCF Files From GATK
Both gVCF Genotyper and Joint Genotyper can process gVCF files written by the GATK variant caller
when using GATK v4.1. To enable this option, set --vc-enable-gatk-acceleration for both gVCF
Genotyper and Joint Genotyper.
Genotyper Options
This section provides information on the options available for each of the genotypers.
gVCF Options
In addition to the standard parameters for the variant caller stage of the DRAGEN host software, the
following parameters are available to create a gVCF:
• --vc-emit-ref-confidence
To enable banded gVCF generation, set to GVCF. To enable base pair resolution gVCF generation,
set to BP_RESOLUTION. Banded gVCF generation is enabled by default and recommended. BP_
Resolution results in large files that slow down any subsequent analysis.
• --vc-gvcf-gq-bands
The --vc-gvcf-gq-bands option defines genotype quality (GQ) bands values. If the GQ values for
positions homozygous to the reference lie within the same band, the positions homozygous to the
reference are grouped into the same block.
• --vc-max-alternate-alleles
The --vc-max-alternate-alleles option specifies the maximum number of ALT alleles that are
output in VCF or gVCF. The default value is 6.
gVCF Genotyper Options
The gVCF Genotyper uses a set of single sample gVCFs to output a multisample VCF that contains one
entry per variant seen in any of the input gVCFs. Genotypes cannot be adjusted using population
information.
gVCF Genotyper can also read gVCF files from an S3 bucket. For gVCF files in a public bucket, URLs
with the prefix s3:// or https:// can be used in --variant or --variant-list. If the bucket requires
authentication, environment variables or config files can be used. See the Samtools website for
information on the htslib AWS S3 plugin.
gVCF Genotyper needs access to the index file for each gVCF input. The URLs for each gVCF and index
file needs to be combined as https://url1.gvcf.gz##idx##https://url2.gvcf.gz.tbi, and
then passed to --variant or --variant-list on the command line.
The following parameters are available for gVCF Genotyper.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
107
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --enable-gvcf-genotyper
To enable the gVCF Genotyper, set to true.
• --ht-reference
The file containing the reference sequence in FASTA format. --ht-reference is required.
• --output-directory
The output directory. --output-directory is required.
• --output-file-prefix
The prefix used to label all output files. --output-file-prefix is required.
• --gg-output-format
The output file format. The default value is vcf.gz. The permitted output file formats are vcf.gz, vcf,
or bcf. Only the vcf.gz format is compatible with the joint genotyper. If using a different format, you
can convert the format using the open-source bcftools utility.
• --gg-regions
The file specifying the regions to run the gVCF Genotyper in. Variants outside these regions are
ignored. The file can either be a bed file or a list of genomic regions specified using
chromosome:start-end. Genomic regions can be separated by commas or line breaks. If using
exome or enrichment data, specify the list of regions targeted by the probes to limit additional time
spent processing unreliable genotype variants that lie outside the targeted regions.
• --gg-enable-concat
Concat output for genomic regions into a single output file. By default, the value is set to true.
• --gg-max-alternate-alleles
Maximum number of alternate alleles. By default, the value is set to 50. If there are more alleles
than the set limit, alleles are ranked by frequency of occurrence in the input samples. The most
common k alleles are output.
• --gg-enable-indexing
Build a tabix index for the output file. By default, the value is set to false. The --gg-output-format
must be set to vcf.gz to use --gg-enable-indexing.
• --gg-extra-params=--min-depth=X
[Optional]Remove false-positive variant calls. Values for genotypes (FORMAT/GT) in samples with
read depth (FORMAT/DP) below the threshold are set to missing. The default threshold is 8.
• --gg-drop-genotypes
Select to output only the alleles for each variant. By default, the value is set to false. --gg-drop-
genotypes is equivalent to running bcftools view -G on the default output.
• --gg-allele-list
[Optional] Force the output of genotypes at specified sites. The path of a vcf.gz or bcf file
containing the sites must be included.
• --gg-extra-params=--remove-nonref
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
108
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
[Optional]Removes the <NON_REF> symbolic allele from the output of gVCF Genotyper. This
option should be used to support downstream tools which cannot process VCF lines with <NON_
REF>.
Joint Genotyper Options
You can run the Joint Genotyper from a multisample VCF, a multisample gVCF, or directly from a set of
single sample gVCFs.
The following parameters are available for Joint Genotyper.
• --enable-joint-genotyping
To run the Joint Genotyper, set to true.
• --output-directory
The output directory. --output-directory is required.
• --output-file-prefix
The prefix used to label all output files. --output-file-prefix is required.
• -r
The directory where the hash table resides.
• --variant or --variant-list
Specifies the path to a single gVCF file. You can specify multiple gVCF files using multiple --variant
options. A maximum of 200 gVCFs are supported. Use --variant-list to specify a file containing a list
of gVCF files that need to be combined using one variant file path per line.
• --pedigree-file
Specify the path to a pedigree file describing the relationship between samples. For more
information, see Pedigree Mode on page 103.
Combine gVCF Options
You can run Combine gVCFs on a set of single sample gVCFs and multisample gVCFs to create a single
multisample gVCF as output. For large-scale population calling, use gVCF Genotyper instead of the
Combine gVCF Genotyper.
The following parameters are available for combine gVCFs:
• --enable-combinegvcfs
To run Combine gVCFS, set to true.
• --intermediate-results-dir
[Optional] Set the intermediate results to a directory different from the output directory, such as
/staging/temp. If running Combine gVCFs, --intermediate-results-dir is recommended. For more
information, see Determine Input and Output File Locations on page 22.
• --output-directory
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
109
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The output directory. --output-directory is required.
• --output-file-prefix
The prefix used to label all output files. --output-file-prefix is required.
• -r
The directory where the hash table resides.
• --variant or --variant-list
Specifies the path to a single gVCF file. You can specify multiple gVCF files using multiple --variant
options. 200 gVCFs are supported, but it is not recommended to combine more than 10 gVCFS. Use
--variant-list to specify a file containing a list of gVCF files that need to be combined using one
variant file path per line.
Joint Analysis Output Format
There are two available joint analysis output files:
• Multisample VCF—A VCF file containing a column with genotype information for each of the input
samples according to the input variants.
• Multisample gVCF—A gVCF file augmenting the content of a multisample VCF file, similar to how a
gVCF file augments a VCF file for a single sample. In between variant sites, the multisample gVCF
contains statistics that describe the level of confidence that each sample is homozygous to the
reference genome. Multisample gVCF is a convenient format for combining the results from a
pedigree or small cohort into a single file. If using a large number of samples, fluctuation in
coverage or variation in any of the input samples creates a new hom-ref block, which causes a
highly fragmented block structure and a large output file that can be slow to create.
Hom-ref Blocks FORMAT Fields
In hom-ref blocks, the following FORMAT fields are calculated uniquely.
• FORMAT/DP—Values represent the minimum DP across all positions within the band.
• FORMAT/AD—Values represent the position in the band where DP=median DP.
• FORMAT/AF—Values are based on FORMAT/AD.
• FORMAT/PL—Values represent the Phred likelihoods per genotype hypothesis. For hom-ref blocks,
each value in FORMAT/PL represents the minimum value across all positions within the band.
• FORMAT/SPL and FORMAT/ICNT—Parameters reported in the gVCF records, including both hom-
ref blocks and variant records. The parameters are used to compute the confidence score of a
variant being de novo in the proband of a trio. For SNP, FORMAT/PL and FORMAT/SPL are both
used as input to the DeNovo caller. FORMAT/PL represents Phred likelihoods obtained from the
genotyper, if the genotyper is called. FORMAT/SPL represents Phred likelihoods obtained from
column-wise estimation, pre-graph. Each value in FORMAT/SPL represents the minimum across all
positions within the band. For INDEL, the PL value is computed in the joint pedigree calling step
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
110
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
based on the FORMAT/ICNT reported in the gVCF file. FORMAT/ICNT consist of two values. The
first value is the number of reads with no indels at the position, and the second value is the number
of reads with indels at the position. Each value in FORMAT/ICNT represent the maximum of the
value across all positions within the band.
In the following example hom-ref block, ICNT provides information on if each sample contains an Indel
at the position of interest. If the proband contains an indel at the position and the ICNT of the parents
does not indicate any read supporting an indel, then the confidence score is high for the proband to
have an indel de novo call at the position.
chr1 10288 . C <NON_REF> . PASS END=10290 GT:AD:DP:GQ:MIN_DP:PL:SPL:ICNT
0/0:131,4:135:69:132:0,69,1035:0,125,255:23,1
chr1 10291 . C
T,<NON_REF> 38.45 PASS
DP=100;MQ=24.72;MQRankSum=0.733;ReadPosRankSum=4.112;FractionInformativeRe
ads=0.600;R2_5P_bias=0.000
GT:AD:AF:DP:F1R2:F2R1:GQ:PL:SPL:ICNT:GP:PRI:SB:MB
0/1:28,32,0:0.533,0.000:60:20,21,0:8,11,0:15:73,0,12,307,157,464:255,0,255
:23,10:3.8452e+01,1.3151e-
01,1.5275e+01,3.0757e+02,1.9173e+02,4.5000e+02:0.00,34.77,37.77,34.77,69.5
4,37.77:4,24,7,25:8,20,14,18
The SPL and ICNT is specific to DRAGEN. The GATK variant caller does not output the SPL and ICNT
values.
De Novo Small Variant Filtering
The filtering step identifies de novo variants calls of the joint calling workflow in regions with ploidy
changes. Since de novo calling can have reduced specificity in regions where at least one of the
pedigree members shows non-diploid genotypes, the de novo variant filtering marks relevant variants
and thus can improve specificity of the call set.
Based on the structural and copy number variant calls of the pedigree, the FORMAT/DN field in the
proband is changed from the original DeNovo value to DeNovoSV or DeNovoCNV if the de novo variant
overlaps with a ploidy-changing SV or CNV, respectively. All other variant details remain unchanged,
and all variants of the input VCF will also be present in the filtered output VCF. Structural or copy
number variants which result in no change of ploidy, such as inversions, are not considered in the
filtering. As an example, a de novo SNV calls in the input VCF
chr1 234710899 . T C 44.74 PASS
AC=1;AF=0.167;AN=6;DP=73;FS=4.720;MQ=250.00;MQRankSum=5.310;QD=1.15;ReadPo
sRankSum=1.366;SOR=0.251
GT:AD:AF:DP:GQ:FT:F1R2:F2R1:PL:GL:GP:PP:DQ:DN
0/1:21,18:0.462:39:48:PASS:14,10:7,8:84,0,50:-8.427,0,-5:4.950e+01,7.041e-
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
111
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
05,5.300e+01:15,0,120:3.2280e-01:DeNovo
0/0:13,0:0.000:11:30:PASS:.:.:0,30,450:.:.:10,0,227
0/0:25,0:0.000:22:60:PASS:.:.:0,60,899:.:.:0,33,227
Overlapping with an SV duplication in the proband, mother or father would be represented in the
filtered output VCF as follows:
chr1 234710899 . T C 44.74 PASS
AC=1;AF=0.167;AN=6;DP=73;FS=4.720;MQ=250.00;MQRankSum=5.310;QD=1.15;ReadPo
sRankSum=1.366;SOR=0.251
GT:AD:AF:DP:GQ:FT:F1R2:F2R1:PL:GL:GP:PP:DQ:DN
0/1:21,18:0.462:39:48:PASS:14,10:7,8:84,0,50:-8.427,0,-5:4.950e+01,7.041e-
05,5.300e+01:15,0,120:3.2280e-01:DeNovoSV
0/0:13,0:0.000:11:30:PASS:.:.:0,30,450:.:.:10,0,227
0/0:25,0:0.000:22:60:PASS:.:.:0,60,899:.:.:0,33,227
The following is an example command line for running the de novo filtering, based on the files returned
by the joint calling workflows:
dragen \
--dn-enable-denovo-filtering true \
--dn-input-joint-vcf <JOINT_SMALL_VARIANT_VCF> \
--dn-output-joint-vcf <OUTPUT_VCF> \
--dn-sv-vcf <JOINT_SV_VCF> \
--dn-cnv-vcf <JOINT_CNV_VCF> \
--enable-map-align false
De Novo Small Variant Filtering Options
The following options are used for de novo variant filtering:
• --dn-input-vcf—Joint small variant VCF from the de novo calling step to be filtered.
• --dn-output-vcf—File location to which the filtered VCF should be written. If not specified, the
input VCF is overwritten.
• --dn-sv-vcf—Joint structural variant VCF from the SV calling step. If omitted, checks with
overlapping structural variants are skipped.
• --dn-cnv-vcf— Joint structural variant VCF from the CNV calling step. If omitted, checks with
overlapping copy number variants are skipped.
Germline Variant Small Hard Filtering
DRAGEN provides post-VCF variant filtering based on annotations present in the VCF records. Default
and non-default variant hard filtering are described below. However, due to the nature of DRAGEN’s
algorithms, which incorporate the hypothesis of correlated errors from within the core of variant caller,
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
112
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
the pipeline has improved capabilities in distinguishing the true variants from noise, and therefore the
dependency on post-VCF filtering is substantially reduced. For this reason, the default post-VCF
filtering in DRAGEN is very simple.
Default Small Variant Hard Filtering
The default filters in the germline pipeline are as follows:
• ##FILTER=<ID=DRAGENSnpHardQUAL,Description="Set if true:QUAL < 10.41">
• ##FILTER=<ID=DRAGENIndelHardQUAL,Description="Set if true:QUAL < 7.83">
• ##FILTER=<ID=LowDepth,Description="Set if true:DP <= 1">
• ##FILTER=<ID=PloidyConflict,Description="Genotype call from variant caller not consistent with
chromosome ploidy">
• ##FILTER=<ID=base_quality,Description="Site filtered because median base quality of alt reads at
this locus does not meet threshold">
• ##FILTER=<ID=lod_fstar,Description="Variant does not meet likelihood threshold (default
threshold is 6.3)">
• DRAGENSnpHardQUAL and DRAGENIndelHardQUAL: For all contigs other than the mitochondrial
contig, the default hard filtering consists of thresholding the QUAL value only. A different default
QUAL threshold value is applied to SNP and INDEL.
• lod_fstar: For the mitochondrial contig, the default hard filtering consists of thresholding the LOD
score only.
• base_quality: For the mitochondrial contig, this filter applies to sites where the median base quality
of alt reads falls below a threshold.
• LowDepth: This filter is applied to all variants call with FORMAT/DP <= 1/.
• PloidyConflict: This filter is applied to all variant calls on chrY of a female subject, if female is
specified on the DRAGEN command line, of if female is detected by the Ploidy Estimator.
Non-Default Small Variant Hard Filtering
DRAGEN supports basic filtering of variant calls as described in the VCF standard. You can apply any
number of filters with the --vc-hard-filter option, which takes a semicolon-delimited list of expressions,
as follows:
<filter ID>:<snp|indel|all>:<list of criteria>,
where the list of criteria is itself a list of expressions, delimited by the || (OR) operator in this format:
<annotation ID> <comparison operator> <value>
The meaning of these expression elements is as follows:
• filterID—The name of the filter, which is entered in the FILTER column of the VCF file for calls that
are filtered by that expression.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
113
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• snp/indel/all—The subset of variant calls to which the expression should be applied.
• annotation ID—The variant call record annotation for which values should be checked for the filter.
Supported annotations include FS, MQ, MQRankSum, QD, and ReadPosRankSum.
• comparison operator—The numeric comparison operator to use for comparing to the specified
filter value. Supported operators include <, ≤, =, ≠, ≥, and >.
For example, the following expression would mark with the label "SNP filter" any SNPs with FS < 2.1 or
with MQ < 100, and would mark with "indel filter" any records with FS < 2.2 or with MQ < 110:
--vc-hard-filter=”SNP filter:snp:FS < 2.1 || MQ < 100; indel
filter:indel:FS < 2.2 || MQ < 110”
This example is for illustration purposes only and is NOT recommended for use with DRAGEN V3
output. Illumina recommends using the default hard filters.
The only supported operation for combining value comparisons is OR, and there is no support for
arithmetic combinations of multiple annotations. More complex expressions may be supported in the
future.
Orientation Bias Filter
The orientation bias filter is designed to reduce noise typically associated with the following:
• Pre-adapter artifacts introduced during genomic library preparation (eg, a combination of heat,
shearing, and metal contaminates can result in the 8-oxoguanine base pairing with either cytosine
or adenine, ultimately leading to G→T transversion mutations during PCR amplification), or
• FFPE (formalin-fixed paraffin-embedded) artifact. FFPE artifacts stem from formaldehyde
deamination of cytosines, which results in C to T transition mutations.
The orientation bias filter can only be used on somatic pipelines. To enable the filter, set the --vc-
enable-orientation-bias-filter option to true. The default is false.
The artifact type to be filtered can be specified with the --vc-orientation-bias-filter-artifacts option.
The default is C/T,G/T, which correspond to OxoG and FFPE artifacts. Valid values include C/T, or G/T,
or C/T,G/T,C/A.
An artifact (or an artifact and its reverse compliment) cannot be listed twice. For example, C/T,G/A is
not valid, because C→G and T→A are reverse compliments.
The orientation bias filter adds the following information.
• ##FORMAT=<ID=F1R2,Number=R,Type=Integer,Description="Count of reads in F1R2 pair
orientation supporting each allele">
• ##FORMAT=<ID=F2R1,Number=R,Type=Integer,Description="Count of reads in F2R1 pair
orientation supporting each allele">
• ##FORMAT=<ID=OBC,Number=1,Type=String,Description="Orientation Bias Filter base context">
• ##FORMAT=<ID=OBPa,Number=1,Type=String,Description="Orientation Bias prior for artifact">
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
114
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• ##FORMAT=<ID=OBParc,Number=1,Type=String,Description="Orientation Bias prior for reverse
compliment artifact">
• ##FORMAT=<ID=OBPsnp,Number=1,Type=String,Description="Orientation Bias prior for real
variant">
dbSNP Annotation
In Germline, Tumor-Normal somatic, or Tumor-Only somatic modes, DRAGEN can look up variant calls
in a dbSNP database and add annotations for any matches that it finds there. To enable the dbSNP
database search, set the --dbsnp option to the full path to the dbSNP database VCF or .vcf.gz file,
which must be sorted in reference order.
For each variant call in the output VCF, if the call matches a database entry for CHROM, POS, REF, and
at least one ALT, then the rsID for the matching database entry is copied to the ID column for that call
in the output VCF. In addition, DRAGEN adds a DB annotation to the INFO field for calls that are found
in the database.
DRAGEN matches variant calls based on the name of the reference sequence/contig, but there is no
additional way to assert that the reference used for constructing the dbSNP is the same as the
reference used for alignment and variant calling. Make sure that the contigs in the selected annotation
database match those in the alignment/variant calling reference.
Panel of Normals VCF
When DRAGEN is used in the Tumor-Normal or Tumor-Only somatic mode, a panel of normals (PON)
VCF can be specified for the purpose of filtering out systematic errors. The PON VCF must be
generated ahead of time and represents a set of variants that were detected by the DRAGEN Somatic
Pipeline when run on normal samples that are not matched to the subject from whom the tumor sample
was taken. The PON VCF may contain several dozen samples. The samples should ideally be normal
samples collected on the same library prep/sequencing instrument, so that if there are systematic
errors that occur during sequencing/library prep, they get captured in the PON VCF.
• --panel-of-normals
Specifies a PONVCF file. When a PON VCF file is used as input and if a somatic variant is found in at
least one sample in the file, it is marked as panel_of_normals in the FILTER column of the output
VCF.
Systematic Noise Filtering
When Local Analysis Software is used in the Tumor-Normal or Tumor-Only somatic mode, a BED file
with site-specific noise level can be specified for the purpose of filtering out sequencing / systematic
noise. The site-specific noise level is used to calculate an AQ score, similar to the Phred-scale. If the AQ
score is smaller than the defined threshold, the variant is filtered as systematic noise. The systematic
noise BED file is built using VCFs that were generated by the Local Analysis Software Somatic Pipeline
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
115
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
when run on normal samples that do not necessarily match to the subject the tumor sample was taken
from. The file might contain several dozen samples, ideally normal samples collected on the same
library prep kit and sequencing system, so that if there are systematic errors that occur during library
prep or sequencing, they are captured in the systematic noise BED file.
The following are the available systematic noise command-line options
• --vc-systematic-noise—Specifies a systematic noise BED file. If a somatic variant does not pass
the AQ threshold, the variant is marked as systematic_noise in the FILTER column of the output
VCF.
• --vc-systematic-noise-filter-threshold—Set the AQ threshold. By default the threshold value for
tumor-normal is 10 and 60 for tumor-only.
Several prebuilt systematic noise files for WGS and WES can be downloaded. For best performance
normal samples collected on the same library prep kit and sequencing system should be used to build
the systematic noise BED file.
Generate Systematic Noise BED File
You can generate systematic noise BED files from normal samples collected using library prep,
sequencing system, and panels. If using panel sequencing, 50 samples are recommended.
To generate a BED file, do as follows.
1. Run DRAGEN somatic tumor-only on normal samples with --vc-systematic-noise set to true to
generate VCF output per normal sample.
2. Build the BED file using the VCFs and the following options.
• --vc-systematic-noise-raw-input-list—List of input VCFs. Enter one VCF per line.
• --vc-systematic-noise-germline-vaf-threshold—Minimum VAF to remove potential germlines
from systematic noise file building. Variants with a VAF greater than the threshold are not
considered systematic noise. The default is none.
If using small panels, the recommended threshold is 0.3.
• --vc-systematic-noise-use-germline-tag—Use DRAGEN internal germline tagging to remove
potential germlines. Mutually exclusive with --vc-systematic-noise-germline-vaf-threshold.
The default is false.
If using WGS or WES, the recommended setting is true.
• --vc-systematic-noise-method—Method to calculate the systematic noise level (noise allele
frequency) across samples. Enter mean to calculate the average noise allele frequency, max to
calculate the maximum, or aggregate to calculate total alleles / total depth per locus across
samples. The default is mean.
If using WGS the recommended setting is max. If using WES, the recommended setting is
aggregate. If using small panels for higher sensitivity, the recommended setting is mean or
aggregate.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
116
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

You can also build systematic noise BED files in the cloud using the BaseSpace Sequence Hub
DRAGEN CNV Baseline Builder App.
Prebuilt Systematic Noise BED Files
The following prebuilt systematic noise files for WGS and WES are available for download on the
Illumina DRAGEN BIo-IT Platform Support Site page.
Pre-built Systematic Noise
File
Comment Number of Normal Samples
WGS_hg38_v1.0_systematic_
noise.bed.gz
WGS hg38 28 Samples, mixture of PCRFree NovaSeq,
PCRFree HiSeqX, TruSeq Nano HiSeqX
WGS_hs37d5_v1.0_
systematic_noise.bed.gz
WGS hs37d5 31 Samples, mixture of PCRFree NovaSeq,
PCRFree HiSeqX, TruSeq Nano HiSeqX
WGS_hg19_v1.0_systematic_
noise.bed.gz
WGS hg19 31 Samples, mixture of PCRFree NovaSeq,
PCRFree HiSeqX, TruSeq Nano HiSeqX
WES_Nextera_IDT_hg38_
v1.0_systematic_noise.bed.gz
Nextera library prep;
IDT exome; hg38
47 Samples
WES_Nextera_IDT_hs37d5_
v1.0_systematic_noise.bed.gz
Nextera library prep;
IDT exome; hs37d5
47 Samples
WES_Nextera_IDT_hg19_v1.0_
systematic_noise.bed.gz
Nextera library prep;
IDT exome; hg19
47 Samples
WES_TruSeq_IDT_hg38_v1.0_
systematic_noise.bed.gz
TruSeq library prep;
IDT exome; hg38
53 Samples
WES_TruSeq_IDT_hs37d5_
v1.0_systematic_noise.bed.gz
TruSeq library prep;
IDT exome; hs37d5
53 Samples
WES_TruSeq_IDT_hg19_v1.0_
systematic_noise.bed.gz
TruSeq library prep;
IDT exome; hg19
53 Samples
Autogenerated MD5SUM for VCF Files
An MD5SUM file is generated automatically for VCF output files. This file is in the same output directory
and has the same name as the VCF output file, but with an .md5sum extension appended. For
example, whole_genome_run_123.vcf.md5sum. The MD5SUM files is a single-line text file that contains
the md5sum of the VCF output file. This md5sum exactly matches the output of the Linux md5sum
command.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
117
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Force Genotyping
DRAGENsupports force genotyping (ForceGT) for Germline SNV variant calling. To use ForceGT, use
the --vc-forcegt-vcf option with a list of small variants to force genotype. The input list of small variants
can be a .vcf or .vcf.gz file.
The current limitations of ForceGT are as follows:
• ForceGT is supported for Germline SNV variant calling in the V3 mode. The V1, V2, and V2+ modes
are not supported.
• ForceGT is not supported for Somatic SNV variant calling.
• ForceGT variants do not propagate through Joint Genotyping.
ForceGT Input
DRAGEN software supports only a single ForceGT VCF input file, which must meet the following
requirements:
• Have the same reference contigs as the VCF used for variant calling.
• Be sorted by reference contig name and position.
• Be normalized (parsimonious and left-aligned).
• Contain no complex variants (variants that require more than one substitution / insertion / deletion
to go from ref allele to alt allele). For example, any variant in the ForceGT VCF similar to the
following results in undefined behavior in the DRAGEN software:
chrX 153592402 GTTGGGGATGCTGAC CACCCTGAAGGG
The following nonnormalized variants cause undefined behavior in DRAGENsoftware:
• Not parsimonious: chrX 153592402 GC GCG
• Parsimonious representation: chrX 153592403 C CG
ForceGT Operation and Expected Outcome
When running Germline SNV variant calling with ForceGT, DRAGEN generates a single sample gVCF
using the ForceGT VCF as input on the command line. The single sample gVCF output file contains all
regular calls and the forceGT calls, as follows:
• For a ForceGT call that was not called by the variant caller (not common), the call is tagged with
FGT in the INFO field.
• For a ForceGT call that was also called by the variant caller and filter field is PASS (common), the
call is tagged with NML:FGT in the INFO field (NML denotes normal)
• For a normal call (and PASS) by the variant caller, with no ForceGT call (normal), no extra tags are
added (no NML tag, no FGT tag)
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
118
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

This scheme distinguishes among calls that are present due to FGT only, common in both ForceGT
input and normal calling, and normal calls.
All the variants in the input ForceGT VCF are genotyped and present in the output single sample gVCF
file. The following table lists the reported GTs for the variants.
Condition Reported GT
At a position with no coverage ./.
At a position with coverage but no reads
supporting ALT allele
0/0
At a position with coverage and reads
supporting ALT allele
0/0 or 0/1 or
1/1 or 1/2
At a position where the variant call made by default DRAGEN software is different from the one
specified in the input ForceGT vcf, there are multiple entries at the same position in the output gVCF,
as follows:
• One entry for the default DRAGEN variant call, and
• One entry each for every variant call present in the input ForceGT VCF at that position.
chrX 100 G C [Default DRAGEN variant call]
chrX 100 G A [Variant in ForceGT vcf]
If a target bed file is provided along with the input ForceGT VCF, then the output gVCF file only
contains ForceGT variants that overlap the bed file positions.
Copy Number Variant Calling
The DRAGEN Copy Number Variant (CNV) pipeline can call CNV events using next-generation
sequencing (NGS) data. This pipeline supports multiple applications in a single interface via the
DRAGEN Software, including processing of whole-genome sequencing data and whole-exome
sequencing data for germline analysis.
The DRAGEN CNV pipeline supports two normalization modes of operation. The two modes apply
different normalization techniques to handle biases that differ based on the application, for example,
WGS versus WES. While the default option settings attempt to provide the best trade-off in terms of
speed and accuracy, a specific workflow may require more finely tuned option settings.
CNV Workflow
The DRAGEN CNV pipeline follows the workflow shown in the following figure.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
119
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Figure 3 DRAGEN CNV Pipeline Workflow
This pipeline uses many aspects of the DRAGEN platform available in other pipelines, such as hardware
acceleration and efficient I/O processing. To enable CNV processing in the DRAGEN Host Software, set
the --enable-cnv command line option to true.
The CNV pipeline has the following processing modules:
• Target Counts—binning of the read counts and other signals from alignments.
• Bias Correction—correction of intrinsic system biases.
• Normalization—detection of normal ploidy levels and normalization of the case sample.
• Segmentation—breakpoint detection via segmentation of the normalized signal.
• Calling / Genotyping—thresholding, scoring, qualifying, and filtering of putative events as copy
number variants.
The normalization module can optionally take in a panel of normals (PoN), which is used when a cohort
or population samples are readily available. All other modules are shared between the different CNV
algorithms.
Signal Flow Analysis
The following figures show a high-level overview of the steps in the DRAGEN CNV Pipeline as the signal
traverses through the various stages. These figures are examples and are not identical to the plots that
are generated from the DRAGEN CNV Pipeline.
The first step in the DRAGEN CNV Pipeline is the target counts stage, which extracts signals such as
read count and improper pairs and puts them into target intervals.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
120
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Figure 4 Read Count Signal
Figure 5 Improper Pairs Signal
Next, the case sample is normalized against the panel of normals, or against the estimated normal
ploidy level, and any other biases are subtracted out of the signal to amplify any event level signals.
Figure 6 Pre/Post Tangent Normalization
The normalized signal is then segmented using one of the available segmentation algorithms, and
events are called from the segments.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
121
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Figure 7 Segments
Figure 8 Called Events
The events are then scored and emitted in the output VCF.
CNV Pipeline Options
The following are the top-level options that are shared with the DRAGEN Host Software to control the
CNV pipeline. The input into the DRAGEN CNV can be a BAM or CRAM file. If you are using the DRAGEN
mapper and aligner, FASTQ files can be used.
• --bam-input—The BAM file to be processed.
• --cram-input—The CRAM file to be processed.
• --enable-cnv—Enable or disable CNV processing. Set to true to enable CNV processing.
• --enable-map-align—Enables the mapper and aligner module. The default is true, so all input reads
are remapped and aligned unless this option is set to false.
• --fastq-file1, --fastq-file2—FASTQ file, or files, to be processed.
• --output-directory—Output directory where all results are stored.
• --output-file-prefix—Output file prefix that will be prepended to all result file names.
• --ref-dir—The DRAGEN reference genome hashtable directory.
CNV Pipeline Input
The DRAGEN CNV pipeline supports multiple input formats. The most common format is an already
mapped and aligned BAM or CRAM file. If you have data that has not yet been mapped and aligned, see
Generate an Alignment File on page 123.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
122
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
To run the DRAGEN CNV Pipeline directly with FASTQ input without generating a BAM or CRAM file,
then see Streaming Alignments on page 124, which outlines steps for streaming alignment records
directly from the DRAGEN map/align stage.
Reference Hashtable
For the DRAGEN CNV pipeline, the hashtable must be generated with the --enable-cnv option set to
true, in addition to any other options required by other pipelines. When --enable-cnv is true, dragen
generates an additional k-mer uniqueness map that the CNV algorithm uses to counteract mapability
biases. The k-mer uniqueness map file only needs to be generated once per reference hashtable and
takes about 1.5 hours per whole human genome.
The reference hashtable is a pregenerated binary representation of the reference genome. For
information on generating a hashtable, see Prepare a Reference Genome on page 8.
The following is an example of a command to generate a hashtable.
dragen \
--build-hash-table true \
--ht-reference <FASTA> \
--output-directory <OUTPUT> \
--enable-cnv true \
--enable-rna true
Generate an Alignment File
The following command line examples show how to run the DRAGEN map/align pipeline depending on
your input type. The map/align pipeline generates an alignment file in the form of a BAM or CRAM file
that can then be used in the DRAGEN CNV Pipeline.
You need to generate alignment files for all samples that have not already been mapped and aligned,
including any samples to be used as references for normalization. Each sample must have a unique
sample identifier, which is specified with the --RGSM option. For BAMand CRAMinput files, the sample
identifier is taken from the file, so the --RGSM option is not required.
Example command to map/align a FASTQ file:
dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
--RGSM <SAMPLE> \
--RGID <RGID> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true
Example command to map/align an existing BAM file:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
123
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
dragen \
-r <HASHTABLE> \
--bam-input <BAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true
Example command to map/align an existing CRAMfile:
dragen \
-r <HASHTABLE> \
--cram-input <CRAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true
Streaming Alignments
DRAGEN can map and align FASTQ samples and then directly stream them to downstream callers.
Examples of downstream callers include the CNV Caller and the Haplotype Variant Caller. This process
allows you to skip generation of a BAM or CRAM file, bypassing the need to store additional files.
To stream alignments directly to the DRAGEN CNV pipeline, run the FASTQ sample through a regular
DRAGEN map/align workflow, and then provide additional arguments to enable CNV. The following
shows an example command line to map/align a FASTQ file and send it to the Germline CNVWGS
pipeline.
dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
--RGSM <SAMPLE> \
--RGID <RGID> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true \
--enable-cnv true \
--cnv-enable-self-normalization true
For information on running CNV concurrently with the Haplotype Variant Caller, see Concurrent CNV
and Small Variant Calling on page 140.
Target Counts
The target counts stage is the first processing stage for the DRAGEN CNV pipeline. This stage bins the
alignments into intervals. The primary analysis format for CNV processing is the target counts file,
which contains the feature signals that are extracted from the alignments to be used in downstream
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
124
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
processing. The binning strategy, interval sizes, and their boundaries are controlled by the target
counts generation options, and the normalization technique used.
When working with whole genome sequence data, the intervals are autogenerated from the reference
hashtable. Only the primary contigs from the reference hashtable are considered for binning. You can
specify additional contigs to bypass with the --cnv-skip-contig-list option.
With whole exome sequence data, the target BED file supplied with the --cnv-target-bed option is used
to determine the intervals for analysis.
The target counts stage generates a .target.counts.gz file, which can be later used in place of any BAM
or CRAM by specifying it with the --cnv-input option for the normalization stage. The .target.counts.gz
file is an intermediate file for the DRAGEN CNV pipeline and should not be modified.
The .target.counts.gz file is a tab-delimited compressed text file with the following columns:
• Contig identifier
• Start position
• End position
• Target interval name
• Count of alignments in this interval
• Count of improperly paired alignments in this interval
An example of a *.target.counts.gz file is shown below.
contig start stop name SampleName
improper_pairs
1 565480 565959 target-wgs-1-565480 7 6
1 566837 567182 target-wgs-1-566837 9 0
1 713984 714455 target-wgs-1-713984 34 4
1 721116 721593 target-wgs-1-721116 47 1
1 724219 724547 target-wgs-1-724219 24 21
1 725166 725544 target-wgs-1-725166 43 12
1 726381 726817 target-wgs-1-726381 47 14
1 753243 753655 target-wgs-1-753243 31 2
1 754322 754594 target-wgs-1-754322 27 0
1 754594 755052 target-wgs-1-754594 41 0
Whole Genome
If the samples are whole genome, then the effective target intervals width is specified with the --cnv-
interval-width option. The higher the coverage of a sample, the higher the resolution that can be
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
125
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

detected. This option is important when running with a panel of normals because all the samples must
have matching intervals. For self normalization, the effective width may be larger than the specified
value.
The default value for WGS is 1000 bp with a sample coverage of ≥ 30x.
WGS Coverage per
Sample
Recommended
Resolution* (bp)
5 10000
10 5000
≥30 1000
*Using a cnv-interval-width of ≤250 bp for WGS analysis can drastically increase run time
The intervals are autogenerated for every contig in the reference. You can specify a list of contigs to
skip by using the --cnv-skip-contig-list option. This option takes comma-separated list of contig
identifiers. The contig identifiers must match the reference hashtable that you are using. By default,
only the mitochondrial chromosomes are skipped. Non-primary contigs are never processed.
For example, to skip chromosome M, X, and Y, use the following option:
--cnv-skip-contig-list "chrM,chrX,chrY"
Whole Exome
If the samples are whole exome samples, a target BED file should be supplied with the --cnv-target-
bed $TARGET_BED option.
The intervals in target BED file indicate regions where alignments are expected based on the target
capture kit. The BED file intervals are further split into intervals of smaller size, depending on the value
of cnv-interval-width.
To use a standard BED file, make sure that there is no header present in the file. In this case, all
columns after the third column are ignored, similar to the operation of DRAGEN Variant Caller.
Target Counts Options
The following options control the generation of target counts.
• --cnv-counts-method—Specifies the counting method for an alignment to be counted in a target
bin. Values are midpoint, start, or overlap. The default value is overlap when using the panel of
normal approach, which means if an alignment overlaps any part of the target bin, it is counted for
that bin. In the self normalization mode, the default counting method is start.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
126
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --cnv-min-mapq—Specifies the minimum MAPQ for an alignment to be counted during target
counts generation. The default value is 3 for self normalization and 20 otherwise. When generating
counts for panel of normals, all MAPQ0 alignments are counted.
• --cnv-target-bed—Specifies a properly formatted BED file that indicates the target intervals to
sample coverage over. For use in WES analysis.
• --cnv-interval-width—Specifies the width of the sampling interval for CNV processing. This option
controls the effective window size. The default is 1000 for WGS analysis and 500 for WES analysis.
• --cnv-skip-contig-list—Specifies a comma-separated list of contig identifiers to skip when
generating intervals for WGS analysis. The default contigs that are skipped, if not specified, are
"chrM,MT,m,chrm".
Target Counts Dropout Regions
In the WGS case where a BED file is not specified for a given reference, the same intervals should be
generated each time. The intervals created take into account the mappability of the reference genome
using a k-mer uniqueness map created during hashtable generation. A dropout region is a complex
region that does not count alignments and results in an interval missing from the analysis. Dropout
regions include centromeres, telomeres, and low complexity regions. If there is sufficient signal in the
flanking regions, an event can still span these dropout regions, even if alignment counting does not
occur in the regions. The event is handled by the segmentation stage.
GC Bias Correction
GC Biases measure the relationship between GC content and read coverage across a genome. Biases
can occur in library prep, capture kits, sequencer differences, and mapping, resulting in difficulties
calling CNV events. The Consumable Prefix GC bias correction module attempts to correct these
biases.
The GC bias correction module immediately follows the target counts stage and operates on the
.target.count file. GC bias correction generates a GC bias corrected version of the file, which has a
.target.counts.gc-corrected.gz extension in the file name. The GC bias corrected versions are
recommended for any downstream processing when working with WGS data. For WES, if there are
enough target regions, then the GC bias corrected counts can also be used.
Typical capture kits have over 200,000 targets spanning the regions of interest. If your BED file has
fewer than 200,000 targets, or if the target regions are localized to a specific region in the genome
(such that GC bias statistics may be skewed), then GC bias correction should be disabled.
The following options control the GC bias correction module.
• --cnv-enable-gcbias-correction—Enable or disable GC bias correction when performing target
counts generation. The default is true.
• --cnv-enable-gcbias-smoothing—Enable or disable smoothing the GC bias correction across
adjacent GC bins with an exponential kernel. The default is true.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
127
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --cnv-num-gc-bins—Specifies the number of bins for GC bias correction. Each bin represents the
GC content percentage. Allowed values are 10, 20, 25, 50, or 100. The default is 25.
Normalization
The DRAGEN CNV pipeline supports two normalization algorithms:
• Self Normalization—Estimates the autosomal diploid level from the sample under analysis to
determine the baseline level to normalize by. Sex chromosomes and PAR regions are handled
based on the sample sex.
• Panel of Normals—A reference-based normalization algorithm that uses additional matched normal
samples to determine a baseline level from which to call CNV events. The matched normal samples
in this case means it has undergone the same library prep and sequencing workflow as the case
sample.
Which algorithm to use depends on the available data and the application. Use the following guidelines
to select the mode of normalization.
Self Normalization
• Whole genome sequencing
• Single sample analysis
• Additional matched samples are not readily available
• Simpler workflow via a single invocation
Panel of Normals
• Whole genome sequencing
• Whole exome sequencing
• Targeted panels, including somatic panels
• Additional matched samples are available
• Tumor/Matched-Normal analysis
• Non-human samples
Self Normalization
The DRAGEN CNV pipeline provides the self normalization mode that does not require a reference
sample or a panel of normals. Enable this mode by setting --cnv-enable-self-normalization to true. This
operating mode bypasses the need to run two stages and can save time. It uses the statistics within
the case sample itself to determine the baseline from which to make a call.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
128
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Because self normalization uses the statistics within the case sample, this mode is not recommended
for WES or targeted sequencing analysis due to the potential for insufficient data.
The self normalization mode is the recommended approach for whole-genome sequencing single
sample processing. The pipeline continues through to the segmentation and calling stage, producing
the final called events.
dragen \
-r <HASHTABLE> \
--bam-input <BAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true \
--cnv-enable-self-normalization true
If you are running from a FASTQ sample, then the default mode of operation is self normalization.
When operating in self normalization mode, the --cnv-interval-width option that is used during the
target counts stage becomes the effective interval width based on the number of unique k-mer
positions. You typically do not have to modify this option.
Self normalization auto-generates the target intervals for use during analysis based on the reference
genome and only works for standard human references. Non-standard human references require a
BED file to process and the Panel of Normals approach.
Panel of Normals
The Panel of Normals approach uses a set of matched normal samples to determine the baseline level
from which to call CNV events. These matched normal samples should be derived from the same library
prep and sequencing workflow that was used for the case sample. This allows the algorithm to subtract
out system level biases that are not sample specific.
In this mode of operation, the DRAGEN CNV pipeline is broken down into two distinct stages. The
target counts stage is performed on each sample, case, and normals, to bin the alignments. The
normalization and call detection stage is then performed with the case sample against the panel of
normals to determine the events.
Target Counts Stage
Target counts should be performed for all your samples, whether they are to be used as references or
are the case samples under investigation. The case sample and all samples to be used as a panel of
normals sample must have identical intervals and therefore should be generated with identical
settings. The target counts stage also performs GC Bias correction, which is enabled by default.
The examples below are for WGS processing. For exome processing, see Whole Exome on page 126.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
129
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The following is an example command for processing a BAM file.
dragen \
-r <HASHTABL> \
--bam-input <BAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE. \
--enable-map-align false \
--enable-cnv true
The following is an example command for processing a CRAM file.
dragen \
-r <HASHTABLE> \
--cram-input <CRAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true
Normalization and Call Detection Stage
The next step in the CNV pipeline when using a panel of normals is to perform the normalization and to
make the calls. This step involves selecting a panel of normals, which is a list of target counts files to be
used for reference-based median normalization.
You can run the analysis in other workflow combinations, keeping in mind that the CNV events are
called for the reference samples used. Ideally the panel of normals samples follow library prep and
sequencing workflows that are identical to the workflows of the case sample under analysis. For calling
on sex chromosomes, it is recommended that you use sex matched references in the panel. Because
the normalization is performed on a per-target basis against the panel of normals median, having sex
matched references is critical to detecting copy number events on the sex chromosomes.
For optimal bias correction, a minimum of 50 samples is recommended as a panel. DRAGEN can run
with a single-sample panel, but single-sample panels can result in artifactual calls in the test sample
where the panel sample has copy number changes.
To generate a Panel of Normals (PON), create a plain text file in which each line in the file contains a
path pointing to a target.counts.gz file generated from the target counts stage. Relative paths are
supported provided the paths are relative to the current working directory. Absolute paths are
recommended in case the workflow is used later or shared with other users.
The following is an example PON file, which uses a subset of the GC corrected files from the target
counts stage.
/data/output_trio1/sample1.target.counts.gc-corrected.gz
/data/output_trio1/sample2.target.counts.gc-corrected.gz
/data/output_trio2/sample4.target.counts.gc-corrected.gz
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
130
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
/data/output_trio2/sample5.target.counts.gc-corrected.gz
/data/output_trio3/sample7.target.counts.gc-corrected.gz
/data/output_trio3/sample8.target.counts.gc-corrected.gz
....
Alternatively, the files to be used in the panel of normals can be specified with the --cnv-normals-file
option. This option takes a single file name, and can be specified multiple times.
After you have created a PON file, you can run the caller by specifying your case sample with the --cnv-
input option and the PON file with the --cnv-normals-list option. Because we recommend using the GC
bias corrected counts from the previous stage, there is no need to run GC bias correction again.
GCbias correction can be disabled by setting --cnv-enable-gcbias-correction to false. For example:
dragen \
-r <HASHTABLE> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true \
--cnv-input <CASE_COUNTS> \
--cnv-normals-list <NORMALS> \
--cnv-enable-gcbias-correction false
This command normalizes the case sample against the panel of normals.
Panels of Normals for Somatic Analysis
For WGS applications, DRAGEN supports somatic analysis for both Tumor Matched Normal mode as
well as Tumor Only mode, including allele specific copy number calling. For more information, see
Somatic CNV Calling on page 146.
For somatic targeted panels, the use of a panel of normals as the reference baseline can provide
insight into copy number variants. The reported events are based solely on the normalized copy ratio
values and the deviation from the expected reference baseline levels. This might be sufficient for some
applications requiring only the detection of gains and losses in targeted genes.
Normalization Options
These options control the preconditioning of the panel of normals and the normalization of the case
sample.
• --cnv-enable-self-normalization—Enable/disable self normalization mode, which does not require
a panel of normals.
• --cnv-extreme-percentile—Specifies the extreme median percentile value at which to filter out
samples. The default is 2.5.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
131
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• --cnv-input—Specifies a target counts file for the case sample under investigation when using a
panel of normals.
• --cnv-normals-file—Specifies a target.counts.gz file to be used in the panel of normals. This option
can be specified multiple times, once for each file.
• --cnv-normals-list—Specifies a text file containing paths to the list of reference target counts files
to be used as a panel of normals. Absolute paths are recommended in case the workflow is used
later or shared with other users. Relative paths are supported provided the paths are relative to the
current working directory.
• --cnv-max-percent-zero-samples—Specifies a threshold for filtering out targets with too many
zero coverage samples. The default is 5%.
• --cnv-max-percent-zero-targets—Specifies a threshold for filtering out samples with too many
zero coverage targets. The default is 2.5%.
• --cnv-target-factor-threshold—Specifies the bottom percentile of panel-of-normals medians to
filter out useable targets. The default is 1% for whole genome processing and 5% for targeted
sequencing processing.
• --cnv-truncate-threshold—Specifies a percentage threshold for truncating extreme outliers. The
default is 0.1%.
Segmentation
After a case sample has been normalized, it goes through a segmentation stage. There are multiple
segmentation algorithms implemented in DRAGEN, including the following:
• CBS (Circular Binary Segmentation)
• SLM (Shifting Level Models)
The SLM algorithm has three variants, SLM, HSLM, and ASLM. HSLM (Heterogeneous SLM) is for use
in exome analysis and handles target capture kits that are not equally spaced. ASLM (Adaptive SLM)
includes additional sample-specific estimation of technical variability of depth of coverage (as opposed
to changes in copy number), based on the median variance within fixed windows or a preliminary set of
segments based on b-allele ratios, and can provide more robustness to "noisy" or "wavy" samples.
The default segmentation algorithm in use is SLM for germline whole genome processing, ASLM for
somatic whole genome processing, and CBS for whole exome processing.
• --cnv-segmentation-mode—Specifies the segmentation algorithm to perform. The default value is
determined by the type of analysis. The following are the available default values.
Analysis Default
Germline WGS SLM
Somatic WGS ASLM
Targeted/WES HSLM
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
132
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --cnv-merge-distance—Specifies the maximum number of base pairs between two segments that
would allow them to be merged. The default value is 0 for WGS, which means the segments must
be directly adjacent. For WES analysis this parameter is disabled by default due to the spacing of
targeted intervals.
• --cnv-merge-threshold—Specifies the maximum segment mean difference at which two adjacent
segments should be merged. The segment mean is represented as a linear copy ratio value. The
default is 0.2 for WGS and 0.4 for WES. To disable merging, set the value to 0.
Circular Binary Segmentation
Circular Binary Segmentation is implemented directly in DRAGEN and is based on A faster circular
binary segmentation for the analysis of array CGH data with enhancements to improve sensitivity for
NGS data.
The following options control Circular Binary Segmentation.
• --c-alpha—Specifies the significance level for the test to accept change points. The default is 0.01.
• --cnv-cbs-eta—Specifies the Type I error rate of the sequential boundary for early stopping when
using the permutation method. The default is 0.05.
• --cnv-cbs-kmax—Specifies maximum width of smaller segment for permutation. The default is 25.
• --cnv-cbs-min-width—Specifies the minimum number of markers for a changed segment. The
default is 2.
• --cnv-cbs-nmin—Specifies the minimum length of data for maximum statistic approximation. The
default is 200.
• --cnv-cbs-nperm—Specifies the number of permutations used for p-value computation. The
default is 10000.
• --cnv-cbs-trim—Specifies the proportion of data to be trimmed for variance calculations. The
default is 0.025.
Shifting Level Models Segmentation
The Shifting Level Models (SLM) segmentation mode follows from the R implementation of SLMSuite: a
suite of algorithms for segmenting genomic profiles.
• --cnv-slm-eta—Baseline probability that the mean process changes its value. The default is 1e-5.
• --cnv-slm-fw—Minimum number of data points for a CNV to be emitted. The default is 0, which
means segments with one design probe could in effect be emitted.
• --cnv-slm-omega—Scaling parameter modulating relative weight between experimental/biological
variance. The default is 0.3.
• --cnv-slm-stepeta—Distance normalization parameter. The default is 10000. This option is only
valid for HSLM.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
133
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Regardless of the segmentation method, initial segments are split across large gaps where depth data
is unavailable, such as across centromeres.
Quality Scoring
Quality scores are computed using a probabilistic model that uses a mixture of heavy tailed probability
distributions (one per integer copy number) with a weighting for event length. Noise variance is
estimated. The output VCF contains a phred-scale metric measuring confidence in called amplification
(CN > 2 for diploid locus), deletion (CN < 2 for diploid locus), or copy neutral (CN=2 for diploid locus)
events.
The scoring algorithm also calculates exact-copy-number quality scores that are inputs to the DeNovo
CNV detection pipeline.
Blacklist Filtering
You can input blacklist BED to the CNV caller to filter out regions from analysis. This is useful if there
are certain regions in the genome, which are known to be problematic. You can also blacklist larger
intervals that specify common CNVs to aid in downstream analysis. DRAGEN does not provide a
prebuilt blacklist, but you can use the cnv-blacklist-bed option to specify the intervals to blacklist. The
intervals should be formatted in standard three-column BED format.
The intervals in the blacklist are compared with the original target counts intervals. If the overlap is
greater than cnv-blacklist-min-overlap, the target counts interval are excluded from analysis. The
*.target.counts.gz file still includes the interval, so you can inspect the original read counts. The
normalization stage removes intervals and is reflected in the *.tn.tsv.gz file.
A blacklist interval does not guarantee that a CNV call does not span the interval. If there is sufficient
data flanking the region, the segmentation stage along with any merging might still generate a call
spanning the blacklisted interval. However, the call would not take read counts from blacklisted
intervals into account.
Output Files
The DRAGEN host software generates many intermediate files. The final call file that contains the
amplification and deletion events is the *.seg.called.merged file.
In addition to the segment file, DRAGEN emits the calls in the standard VCF format. By default, the VCF
file includes only copy number gain and loss events. For copy neutral segments, refer to the
*.seg.called.merged file. To have copy neutral (REF) calls included in the output VCF, set --cnv-enable-
ref-calls to true.
For more information about the .seg.called.merged file, and how to use the output files to aid in debug
and analysis, see Signal Flow Analysis on page 120.
CNV VCF File
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
134
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The CNV VCF file follows the standard VCF format. Due to the nature of how CNV events are
represented versus how structural variants are represented, not all fields are applicable. In general, if
more information is available about an event, then it is annotated. Some fields in the DRAGEN CNV VCF
are unique to CNVs.
The following is an example of the header lines that are specific to CNV.
##fileformat=VCFv4.2
##CoverageUniformity=0.402517
##contig=<ID=1,length=249250621>
##contig=<ID=2,length=243199373>
##contig=<ID=3,length=198022430>
##contig=<ID=4,length=191154276>
##contig=<ID=5,length=180915260>
…
##reference=file:///reference_genomes/Hsapiens/hs37d5/DRAGEN
##ALT=<ID=CNV,Description="Copy number variant region">
##ALT=<ID=DEL,Description="Deletion relative to the reference">
##ALT=<ID=DUP,Description="Region of elevated copy number
relative to the reference">
##INFO=<ID=REFLEN,Number=1,Type=Integer,Description="Number of
REF positions included in this record">
##INFO=<ID=SVLEN,Number=.,Type=Integer,Description="Difference
in length between REF and ALT alleles">
##INFO=<ID=SVTYPE,Number=1,Type=String,Description="Type of
structural variant">
##INFO=<ID=END,Number=1,Type=Integer,Description="End position
of the variant described in this record">
##INFO=<ID=CIPOS,Number=2,Type=Integer,Description="Confidence
interval around POS">
##INFO=<ID=CIEND,Number=2,Type=Integer,Description="Confidence
interval around END">
##FILTER=<ID=cnvQual,Description="CNV with quality below 10">
##FILTER=<ID=cnvCopyRatio,Description="CNV with copy ratio
within +/- 0.2 of 1.0">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=SM,Number=1,Type=Float,Description="Linear copy
ratio of the segment mean">
##FORMAT=<ID=CN,Number=1,Type=Integer,Description="Estimated
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
135
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

copy number">
##FORMAT=<ID=BC,Number=1,Type=Integer,Description="Number of
bins in the region">
##FORMAT=<ID=PE,Number=2,Type=Integer,Description="Number of
improperly paired end reads at start and stop breakpoints">
The ID column is used to represent the event.
The REF column contains an N for all CNV events.
The ALT column specifies the type of CNV event. Because the VCF contains only CNV events, only the
DEL or DUP entry is used.
The QUAL column contains an estimated quality score for the CNV event, which is used in hard filtering.
The FILTER column contains PASS if the CNV event passes all filters, otherwise it contains the name of
the failed filter.
The INFO column contains information representing the event, mostly identical to the ID column. The
REFLEN entry indicates the length of the event. The SVTYPE entry is always CNV. The END entry
indicates the end position of the event. The CIPOS and CIEND entries are currently not used.
The FORMAT fields are described in the header.
• GT—Genotype
• SM—Linear copy ratio of the segment mean
• CN—Estimated copy number
• BC—Number of bins in the region
• PE—Number of improperly paired end reads at start and stop breakpoints
Since germline copy number calling determines overall copy number rather than the copy number on
each haplotype, the genotype type field contains missing values for diploid regions when CN is greater
than or equal to 2. The following are examples of the GT field for various VCF entries:
Diploid or
Haploid?
ALT FORMAT:CN FORMAT:GT
Diploid . 2 ./.
Diploid <DUP> > 2 ./1
Diploid <DEL> 1 0/1
Diploid <DEL> 0 1/1
Haploid . 1 0
Haploid <DUP> > 1 1
Haploid <DEL> 0 1
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
136
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Coverage Uniformity
The DRAGEN CNV pipeline provides a measure of the quality of the data for a sample. The CNV
pipeline assumes that post-normalization target counts are independently and identically distributed
(IID). Coverage in most high-quality WGS samples is uniform enough for the CNV caller to produce
accurate calls, but some samples violate the IID assumption in a manner and to a degree that leads to
unusually high numbers of false positive calls. For such samples, there are local correlations of
coverage bias extending across multiple target intervals so that most or all the target counts in a given
genomic region will be artifactually elevated (or, alternatively, depressed), and the strength of the bias
is sufficient for the region to be identified as a distinct copy number segment and assigned an incorrect
copy number. The CoverageUniformity metric quantifies the degree of local coverage correlation in
the sample to help identify poor-quality samples. CoverageUniformity is present in the VCF header
when WGS self-normalization method is selected. This metric is only available for germline samples.
A larger value for this metric means the coverage in a sample is less uniform, which indicates that the
sample has more nonrandom noise, and could be considered poor quality. The CoverageUniformity
metric depends on factors other than sample quality, such as the cnv-interval-width setting and
sample mean coverage. DRAGEN recommends using this score to compare the quality of samples from
similar mean coverage and the same command line options. Because of this, DRAGEN CNV only
provides the metric and does not take any action based on it.
Visualization and BigWig Files
To perform analysis on a known truth set, you can use the intermediate output files from the pipeline
stages. These files can be parsed to aid in fine-tuning options.
All files have a structure similar to a BED file, with an optional header line.
• *.target.counts.gz—Contains the number of read counts per target interval. This is the raw signal
as extracted from the alignments of the BAM or CRAM file. The format is identical for both the case
sample and any panel of normals samples. There is also a bigWig representation of a
target.counts.diploid file, which is normalized to the normal ploidy level of 2 instead of raw counts.
• *.tn.tsv.gz—The case sample’s tangent normalized signal, per target interval. This file contains the
log-normalized copy ratio signal. A strong signal deviation from 0.0 indicates a potential for a CNV
event.
• *.seg.called.merged—Contains the segments produced from the segmentation algorithm.
• *.cnv.vcf—Output CNV VCF file indicating events.
To generate additional equivalent bigWig and gff files, set the --enable-cnv-tracks option to true.
These files can be loaded into IGV along with other tracks that are available, such as RefSeq genes.
Using these tracks alongside publicly available tracks allows for easier interpretation of calls. DRAGEN
autogenerates IGV session XML file if tracks are generated by DRAGEN CNV. The *.cnv.igv_
session.xml can be loaded directly into IGV for analysis.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
137
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

The following IGV tracks are automatically populated in the output IGV session file:
• .target.counts.bw—BigWig representation of the target counts bins. Setting the track view in IGV to
barchart or points is recommended.
• *.improper_pairs.bw—Bigwig representation of the improper pairs counts. Setting the track view in
IGV to barchart is recommended.
• *.tn.bw—Bigwig representation of the tangent normalized signal. Setting the track view in IGV to
points is recommended.
• *.seg.bw—Bigwig representation of the segments. Setting the track view in IGV to points is
recommended.
• *.cnv.gff3—GFF3 representation of the CNV events. DEL events show up as blue and DUP events
show as red. Filtered events are a light gray. Selecting an event brings up a window for viewing
annotation details.
Figure 9 IGVExample
Exclude Interval Files
To improve accuracy, the DRAGEN CNV Pipeline excludes genomic intervals if one or more the target
intervals failed at least one quality requirement. The excluded intervals are reported to *.excluded_
intervals.bed.gz file. The file identifies the regions of the genome that are not callable for CNV analysis
and describes the reason intervals were excluded in the fourth column. The following are the possible
reasons for exclusion.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
138
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Exclusion Reason Description Command Line Option
NON_KMER_
UNIQUE
Nonunique Kmer bases are larger than
50% of interval.
Not applicable. This reason only
applies to self-normalization
mode.
BLACKLIST Interval overlaps with blacklist larger
than threshold.
--cnv-blacklist-min-overlap
PON_MAX_
PERCENT_ZERO_
SAMPLES
Number of PON samples with 0 coverage
is larger than threshold.
--cnv-max-percent-zero-targets
PON_TARGET_
FACTOR_
THRESHOLD
Median coverage of interval is lower than
threshold of overall median coverage.
--cnv-target-factor-threshold
PON_MISSING_
INTERVAL
Target interval not found in PON Not applicable.
Output and Filtering Options
The output and filtering options control the CNV output files.
• --cnv-blacklist-bed—Specifies a BED file indicating intervals to exclude from the from CNV analysis.
If a target interval overlaps regions specified from blacklist BED file more thancnv-blacklist-min-
overlap, it is suppressed.
• --cnv-blacklist-min-overlap—Specifies a fraction for filtering threshold of overlap amount between
target interval and blacklist region (0.5).
• --cnv-enable-plots—Generate plots as part of the CNV pipeline. The default is false.
• If you perform WGS CNV analysis with high-resolution intervals (less than 1000bp), then plot
generation can take longer to complete. Illumina recommends that you use the default (disabled).
• --cnv-enable-ref-calls—Emit copy neutral (REF) calls in the output VCF file. The default is true for
single WGS CNV analysis.
• --cnv-enable-tracks—Generate track files that can be imported into IGV for viewing. When this
option is enabled, a *.gff file for the output variant calls is generated, as well as *.bw files for the
tangent normalized signal. The default is true.
• --cnv-filter-bin-support-ratio—Filters out a candidate event if the span of supporting bins is less
than the specified ratio with respect to the overall event length. The default ratio is 0.2 (20%
support). As an example, if an event is called and has a length of 100,000 bp, but the target interval
bins that support the call only spans a total of 15,000 bp (15,000/100,000 = 0.15), then it will be
filtered out.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
139
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --cnv-filter-copy-ratio—Specifies the minimum copy ratio threshold value centered about 1.0 at
which a reported event is marked as PASS in the output VCF file. The default value is 0.2, leading to
calls less than CR=0.8 or greater than CR=1.2.
• --cnv-filter-length—Specifies the minimum event length in bases at which a reported event is
marked as PASS in the output VCF file. The default is 10000.
• --cnv-filter-qual—Specifies the QUAL value at which a reported event is marked as PASS in the
output VCF file. The default is 10.
• --cnv-min-qual—Specifies the minimum reported QUAL. The default is 3.
• --cnv-max-qual—Specifies the maximum reported QUAL. The default is 200.
• --cnv-ploidy—Specifies the normal ploidy value. This option is used only for estimation of the copy
number value emitted in the output VCF file. The default is 2.
• --cnv-qual-length-scale—Specifies the bias weighting factor to adjust QUAL estimates for
segments with longer lengths. This is an advanced option and should not need to be modified. The
default is 0.9303 (2-0.1).
• --cnv-qual-noise-scale—Specifies the bias weighting factor to adjust QUAL estimates based on
sample variance. This is an advanced option and should not need to be modified. The default is 1.0.
Concurrent CNV and Small Variant Calling
DRAGEN can perform mapping and aligning of FASTQ samples and then directly stream the data to
downstream callers. A single sample can run through both the CNV and the small VC if the input is a
FASTQ sample. This triggers self normalization by default.
Run the FASTQ sample through a regular DRAGEN map/align workflow, and then provide additional
arguments to either enable the CNV or VC, or both. The options that apply to CNV in the standalone
workflows are also applicable here.
The following examples show different commands.
Map/align FASTQ with CNV
dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
--RGSM <SAMPLE> \
--RGID <RGID> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true \
--enable-cnv true \
--cnv-enable-self-normalization true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
140
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Map/Align FASTQ w/ VC
dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
--RGSM <SAMPLE> \
--RGID <RGID> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true \
--enable-variant-caller true
Map/Align FASTQ w/ CNV and VC
dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
--RGSM <SAMPLE> \
--RGID <RGID> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align true \
--enable-cnv true \
--cnv-enable-self-normalization true \
--enable-variant-caller true
BAM Input to CNV and VC
dragen \
-r <HASHTABLE> \
--bam-input <BAM> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true \
--cnv-enable-self-normalization true \
--enable-variant-caller true
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
141
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Sample Correlation and Sex Genotyper
When running the target counts stage or the normalization stage, the DRAGEN CNV pipeline also
provides the following information about the samples in the run.
• A correlation metric of the read count profile between the case sample and any panel of normals
samples. A correlation metric greater than 0.90 is recommended for confident analysis, but there is
no hard restriction enforced by the software.
• The predicted sex of each sample in the run. The sex is predicted based on the read count
information in the sex chromosomes and the autosomal chromosomes. The median value for the
counts is printed to the screen for the autosomal chromosomes, the X chromosome, and the Y
chromosome.
The results are printed to the screen when running the pipeline. For example:
=============================================
Correlation Table
=============================================
Correlation of case sample PlatinumGenomes_50X_NA12877 against
PlatinumGenomes_50X_NA12878: 0.984092
Sex Genotyper
=============================================
Predicted sex of samples
PlatinumGenomes_50X_NA12877: MALE XY 0.99737
PlatinumGenomes_50X_NA12878: FEMALE XX 0.968929
The predicted sexes for samples in use are also printed to the \*.cnv_metrics.csvoutput file.
To perform analysis on the sex chromosomes using a panel of normals, it is recommended that you use
sex matched samples in the panel of normals.
You can override the sex of the sample with the –sample-sex option .
Multisample CNV Calling
Multisample CNV calling is possible starting from tangent normalized counts files (*.tn.tsv.gz) specified
with the --cnv-input option (one per sample). Multisample CNV analysis benefits from using joint
segmentation to increase the sensitivity of detection of copy number variable segments. For each copy
number variable segment identified, the copy number genotype of each sample is emitted in a single
VCF entry to facilitate annotation and interpretation.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
142
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Multisample CNV calling is recommended for only WGS analysis.
The following is an example command line for running a trio analysis:
dragen \
-r <HASHTABLE> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-cnv true \
--cnv-input <FATHER_TN_TSV> \
--cnv-input <MOTHER_TN_TSV> \
--cnv-input <PROBAND_TN_TSV> \
--pedigree-file <PEDIGREE_FILE>
De Novo CNV Calling Options
All input samples should have gone through the same single sample WGS workflow and must have
identical intervals.
The following options are used in DeNovo CNV calling:
• --cnv-input—For DeNovo CNV calling, this specifies the input tangent-normalized signal files
(*.tn.tsv) from the single sample runs. This option can be specified multiple times, once for each
input sample.
• --cnv-filter-de-novo-qual—Phred-scaled threshold at which a putative event in the proband
sample is marked as DeNovo. Default value is 0.10.
• --pedigree-file—Pedigree file specifying the relationship between the input samples.
Joint Segmentation
First, CNV calling is performed on each sample independently. Joint segmentation then uses the copy
number variable segments from each single sample analysis to derive a set of joint copy number
variable segments. This set of joint segments is determined simply by taking the union of all
breakpoints from the copy number variable segments of all samples. This results in the splitting of any
partially overlapping segments across different samples.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
143
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Figure 10 Overlapping Segments
Following joint segmentation, copy number calling is again performed independently on each sample
using the joint segments. Segments can be merged as with the single sample analysis, but each joint
segment is emitted in the mutlisample VCF as a single entry. The quality score (QS in the VCF) from the
sample's merged segment, if applicable, is used for filtering the call. Sample calls are filtered using the
sample's FT field in the multisample VCF. The QUAL column of the multisample VCF is always missing
(ie, "."). The FILTER column of the mutlisample VCF is "SampleFT" if none of the sample's FT fields are
"PASS", and "PASS" if any of the sample's FT fields are "PASS".
De Novo Calling Stage
A de novo event is defined as the existence of a genotype at a particular locus in a proband’s genome
that did not result from standard Mendelian inheritance from the parents. The denovo calling stage
identifies putative de novo events in the proband of each trio of a multisample analysis. In some cases,
these putative de novo events may be real, but they can also arise from sequencing or analysis
artifacts. Consequently, a de novo quality score is assigned to each putative de novo event and used to
filter out low-quality de novo events. Trios are specified by specifying a .ped file with the --pedigree-file
option. Multiple trios can be specified (eg, quad analysis), and all valid trios will be processed.
For each joint segment in a trio, the de novo caller determines if there is a Mendelian inheritance
conflict for the called copy number genotypes. The CNV caller does not identify the copy number for
each allele of a given diploid segment, which means assumptions are made about the possible allelic
composition of the parent genotypes.
The assumption is that the copy number 0 allele is not present for diploid regions of a parent's genome
(sex dependent) when the assigned copy number is 2 or greater. This results in simplifications, as
follows:
Parent Copy Number
Genotype
Possible Copy Number
Alleles
Assumed Possible Copy Number
Alleles
2 0/2, 1/1 1/1
3 0/3, 1/2 1/2
4 0/4, 1/3, 2/2 1/3, 2/2
N x/(N-x) for x <= N/2 x/(N-x) for 1 <= x <= N/2
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
144
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

The following are examples of consistent and inconsistent copy number genotypes for diploid regions
using these assumptions:
Mother Copy Number Father Copy Number Proband Copy Number Mendelian Consistent?
2 2 2 Yes
2 2 1 No
3 2 4 No
3 2 2 Yes
2 0 2 No
If a joint segment has a Mendelian inheritance conflict, a Phred-scaled de novo quality score (DQ field in
the VCF) is calculated using the likelihoods for each copy number state (see Quality Scoring section) of
each sample in the trio, combined with a prior for the trio genotypes:
DQ = -10log(1-Sum over conflicting genotypes(p(CNm|data)*p(CNf|data)*p
(CNp|data)*p(CNm,CNf,CNp))/Sum over all genotypes(p(CNm|data)*p(CNf|data)*p
(CNp|data)*p(CNm,CNf,CNp)))
Where
• CNm = Mother copy number
• CNf = Father copy number
• CNp = Proband copy number
• p(CNm,CNf,CNp) = the prior for the trio genotype
The DN field in the VCF is used to indicate the de novo status for each segment. Possible values are:
• Inherited - the called trio genotype is consistent with Mendelian inheritance
• LowDQ - the called trio genotype is inconsistent with Mendelian inheritance and DQ is less than the
de novo quality threshold (default 0.1)
• DeNovo - the called trio genotype is inconsistent with Mendelian inheritance and DQ is greater than
or equal to the de novo quality threshold (default 0.1)
Multisample CNV VCF Output
The records in a multisample CNV VCF differ slightly from the single sample case. The major
differences are as follows:
• The per-record entries are broken down into the segments among the union of all the input
samples breakpoints, which means there are more entries in the overall VCF.
• The QUAL column is not used and its value is “.”. The per-sample quality is carried over into the
SAMPLE columns with the QS tag.
• The FILTER column indicates PASS if any of the individual SAMPLE columns PASS. Otherwise, it
indicates SampleFT.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
145
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• The per-sample annotations are carried over from their originating calls. The single sample filters
are applied at the sample level and are emitted in the FT annotation.
Additionally, if a valid pedigree is used, then de novo calling is performed, which adds the following two
annotations to the proband sample.
##FORMAT=<ID=DQ,Number=1,Type=Float,Description="De novo quality">
##FORMAT=<ID=DN,Number=1,Type=String,Description="Possible values are
‘Inherited’, ‘DeNovo’ or ‘LowDQ’. Threshold for a passing de novo call is
DQ > 0.100000">
While the VCF contains many entries, due to the joint segmentation stage, the number of de novo
events can be found by extracting entries that have a DN and DQ annotation. These records are also
extracted and are converted to GFF3 in the de novo calling case.
Somatic CNV Calling
To detect somatic copy number aberrations and regions with loss of heterozygosity, run the DRAGEN
CNV caller on a tumor sample in combination with a VCF containing germline SNVs. The output file is a
VCF file. Components of the germline CNV caller are reused in the somatic algorithm with the addition
of a somatic modeling component, which estimates tumor purity and ploidy.
Figure 11 Somatic CNV Caller Workflow
The germline SNVs are used to compute b-allele ratios in the tumor, which allows for allele-specific
copy number calling on the tumor sample. Where possible, use of the small-variant VCF from a
matched normal sample is preferred ("tumor/normal" mode), but a catalog of population SNPs can be
used when a matched normal sample is not available ("tumor-only" mode).
When a matched normal sample is available, it should first be processed using the germline small
variant caller. In this case, only germline-heterozygous SNV sites are used for determining b-allele
ratios. If no matched normal is available, population SNP b-allele ratios are computed as for matched
normal heterozygous loci, but are treated as variants of unknown germline genotype; possible
genotype assignments are statistically integrated to determine allele specific copy number.
In matched normal mode, a VCF containing germline copy number changes for the individual may
optionally be input. This ensures that germline CNVs are output as separate segments in the somatic
CNV VCF, and annotated with the germline copy number so that it is clear whether there are
specifically-somatic copy number changes in the region.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
146
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Somatic CNV Calling Options
• --tumor-bam-input or --tumor-cram-input
• --cnv-normal-b-allele-vcf or --cnv-population-b-allele-vcf
• --sample-sex
• --cnv-normal-cnv-vcf
• --cnv-use-somatic-vc-vaf
To trigger the somatic CNV caller, the input alignments must use the tumor equivalent options, such as
--tumor-bam-input or --tumor-cram-input. Support for running from FASTQ input (--tumor-fastq1, --
tumor-fastq2, and --tumor-fastq-list) is not available in the somatic CNV caller. In addition, the somatic
CNV caller does not support running directly from the mapper/aligner.
Use the --cnv-normal-b-allele-vcf option to specify a matched normal SNV VCF or --cnv-population-b-
allele-vcf to specify a population SNP catalog. For more information on specifying b-allele loci, see
Specify B-Allele Loci on page 148
If the sample sex is known, use the --sample-sex option to specify the sex. If it is not specified, the
caller attempts to estimate the sample sex from the tumor alignments.
Use --cnv-normal-cnv-vcf to specify germline CNVs from a matched normal sample.
Use --cnv-use-somatic-vc-vaf to control whether the variant allele frequencies (VAFs) or somatic
SNVs are used to help select the tumor model for the sample. For more information, see VAF Aware
Mode on page 149
The following is an example command line for running the minimalsomatic CNV caller.
dragen \
-r <HASHTABLE> \
--output-directory <OUTPUT> \
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true \
--tumor-bam-input <TUMOR_BAM> \
--cnv-normal-b-allele-vcf <SNV_NORMAL_VCF> \
--sample-sex <SEX>
You can enable additional features when a matched-normal sample is also available. If a matched-
normal sample is available, enable germline-aware mode and VAF-aware mode using the following
example command-line. For more information on germline-aware mode and VAF-aware mode, see
Germline-aware Mode on page 149 and VAF Aware Mode on page 149.
dragen \
-r <HASHTABLE> \
--output-directory <OUTPUT> \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
147
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-file-prefix <SAMPLE> \
--enable-map-align false \
--enable-cnv true \
--tumor-bam-input <TUMOR_BAM> \
--cnv-normal-b-allele-vcf <SNV_NORMAL_VCF> \
--enable-vc true \
--bam-input <NORMAL_BAM>
--cnv-normal-cnv-vcf <CNV_NORMAL_VCF> \
--sample-sex <SEX>
If you provide an unmatched normal in place of a matched normal, you need to disable CNV calling or
run in tumor-only mode. You can force tumor-only mode by omitting --cnv-normal-cnv-vcf, specifying -
-cnv-population-b-allele-vcf instead of --cnv-normal-b-allele-vcf, and adding --cnv-use-somatic-vc-
vaf=false.
Target Counts and B-allele Counts
The target counting stage and its output are the same for the germline CNV calling case. The target
intervals with the read counts are output in a *.target.counts file. The b-allele counting occurs in
parallel with the read counting phase, and the values are output in a *.baf.bedgraph.gz file. This file can
be loaded into IGV along with other bigwig files generated by DRAGEN for visualization.
Specify B-Allele Loci
Use the --cnv-normal-b-allele-vcf option to specify a matched normal SNV VCF. Ideally, this VCF file
comes from processing the matched normal sample through the DRAGEN germline small variant caller
with filters applied. Typically, this file name has a *.hard-filtered.vcf.gz extension. All records marked as
PASS that are determined to be heterozygous in the normal sample are used to measure the b-allele
counts of the tumor sample . You can also use equivalent gVCF file (*.hard-filtered.gvcf.gz), but the
processing time is significantly longer due to the number of records, most of which are not
heterozygous sites. It is recommended that you use the VCF file.
Use the --cnv-population-b-allele-vcf option to specify a population SNP VCF. To obtain a population
SNP VCF, process an appropriate catalog of population variation, such as from dbSNP, the 1000G
project, or other large cohort discovery efforts. Only high-frequency SNPs should be included. For
example, include SNPs with minor allele population frequency ≥ 10% to limit run time impact and reduce
artifacts. Specify the ALT allele frequency by adding AF=<alt frequency> to the INFO section of each
record. Additional INFO fields might be present, but DRAGEN only parses and uses the AF field. Sites
specified with --cnv-population-b-allele-vcf can be either heterozygous or homozygous in the germline
genome from which the tumor genome derives.
The following is an example valid population SNP record:
chr1 51479 . T A 1000 PASS AF=0.3253
DRAGEN considers the following requirements when parsing records from the b-allele VCF:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
148
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Only simple SNV sites.
• Records must be marked PASS in the FILTER field.
• If there are duplicate records (same CHROM and POS) in the VCF, then the first occurring record is
used.
Germline-aware Mode
To specify germline CNVs from a matched normal sample, use --cnv-normal-cnv-vcf . When
specified, CNV records marked as PASS in the normal sample are used during tumor-sample
segmentation to ensure that confident germline CNV boundaries are also boundaries in the somatic
output. Segments with germline copy number changes relative to reference ploidy are excluded from
somatic model selection.
During somatic copy number calling and scoring, the germline copy number is used to modify the
expected depth contribution from the normal contamination fraction of the tumor sample, leading to
more accurate assignment of somatic copy number in regions of germline CNV. Finally, somatic CNV
VCF entries are annotated with germline copy number (NCN) and the somatic copy number difference
relative to germline (SCND) for those segments that have germline CNVs.
VAF Aware Mode
When both the small variant caller and the CNV caller are enabled in a tumor-matched normal run, the
somatic SNV results can affect the estimated purity and ploidy of the tumor sample. The somatic SNV
variant allele frequencies (VAFs), as captured by the allele depth values from passing somatic SNVs,
reflect the combination of tumor purity, total tumor copy number at a somatic SNV locus, and the
number of tumor copies bearing the somatic allele. Clusters of somatic SNVs with similar allele depths
inform the tumor model.
Somatic SNV variant allele frequency (VAF) information is especially useful when a tumor has limited
copy number variation and/or CNVs are mostly subclonal, as, for instance, in many liquid tumors.
Without VAF information, the estimated tumor model may be incorrect or low-confidence, leading
either to wrong or filtered calls. VAF information can also help determine the presence or absence of a
genome duplication even in clear, clonal CNVs.
To take advantage of VAF signal, you must run somatic CNV calling with small variant calling on tumor
and matched-normal read and alignment inputs. For example, you could use the following options: --
enable-vc=true --enable-cnv=true --tumor-bam-input T_BAM --bam-input N_BAM
together. By default, VAF-based modeling is enabled. You can disable VAF-based modeling by setting
--cnv-use-somatic-vc-vaf to false.
Somatic CNV Model
Selecting a tumor purity and diploid coverage level (ploidy) is a key component of the somatic CNV
caller. It attempts to fit the observed data, the read counts and b-allele counts, across all segments in
the tumor sample, using a grid-search approach that evaluates many candidate models. A log
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
149
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
likelihood score is emitted for each candidate. These scores are output in the
*.cnv.purity.coverage.models.tsv file. The somatic CNV caller chooses the (purity, coverage) pair with
the highest log likelihood, and computes several measures of model confidence based on the relative
likelihood of the chosen model vs alternative models.
If the confidence in the chosen model is low, then the output VCF marks all records with a
lowModelConfidence FILTER.
Somatic CNV Smoothing
The segmentation stage might produce adjacent or nearby segments that are assigned the same copy
number and have similar depth and BAF data. This can result in fragmentation of a region with
consistent true copy number into several pieces, which may be undesirable for downstream use of
copy number estimates. Additionally, for some uses, it may be preferable to smooth short segments
that would be assigned different copy number whether due to a true copy number change or an
artifact. To reduce undesirable fragmentation, initial segments may be merged during a post-calling
segment smoothing step.
After initial calling, segments shorter than the specified value of --cnv-filter-length are deemed
negligible. Among the remaining non-negligible segments, successive pairs are evaluated for merging.
Two successive segments that are within --cnv-merge-distance of one another and have the same
CN and MCN assignments are combined, along with any intervening neglibible segments, on a trial
basis, into a single segment that is recalled and rescored. If the merged segment receives the same CN
and MCN as its constituent non-neglible pieces, with a sufficiently high quality score, the original
segments are replaced with the merged segment. The merged segment might be further merged with
other initial or merged segments to either side. Merging proceeds until there are not any segment pairs
that meet the merging criteria.
Somatic CNV VCF Output
The somatic CNV VCF file follows the standard VCF format and has the following differences from the
germline CNV VCF output.
The following header lines are specific to somatic CNV calling.
• [ModelSoure]—The primary basis on which the final tumor model was chosen. The following values
are included:
– [DEPTH+BAF]—Depth+BAF signal is used to determine tumor model.
– [DEPTH+BAF_DOUBLED]—The initial depth+BAF model is additionally duplicated based on
VAF signal or excess segments at half the expected depth change.
– [DEPTH+BAF_DEDUPLICATED]—The depth+BAF model is deduplicated based on VAF signal
or insufficient segments supporting a duplication.
– [DEPTH+BAF_WEAK]—Depth+BAF signal is used to determine (lower-confidence) tumor
model.
– [VAF]—VAF signal is used to determine tumor model due to insufficient depth+BAF signal.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
150
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
– [DEGENERATE_DIPLOID]—Sample is treated as high-purity diploid in absence of adequate
signal from depth+BAF and VAF. The diploid coverage is set to lowest value observed in a
substantial number of bases in segments with BAF=50%. All VCF records have
lowModelConfidence added to FILTER value.
– [SAMPLE_MEDIAN]—Sample is treated as high-purity diploid in absence of adequate signal
from depth+BAF and VAF. Diploid coverage set to sample median. All VCF recordsl have
lowModelConfidence added to FILTER value.
• EstimatedTumorPurity—Estimated fraction of cells in the sample due to tumor. The range of this
field is [0, 1].
• DiploidCoverage—Expected read count for a target bin in a diploid region. The numeric value is
unlimited.
• OverallPloidy—Length weighted average of tumor copy number for PASS events. The numeric
value is unlimited.
• AlternativeModelDedup—An alternative to the best model corresponding to one less whole-
genome duplication is given as a pair of values (tumor purity, diploid coverage). This may be useful
for manual investigation where the best model may involve a spurious genome duplication.
• AlternativeModelDup—An alternative to the best model corresponding to one more whole-
genome duplication is given as a pair of values (tumor purity, diploid coverage). This may be useful
for manual investigation where the best model may have missed a true genome duplication.
The ID column represents the type of event. In addition to representing GAIN, LOSS, and REF events,
the CNLOH (copy neutral loss of heterozygosity) and GAINLOH (copy number gain with LOH) entries
represent LOH (loss of heterozygosity) events.
The ALT field may have two alleles, such as <DEL>,<DUP>, which allows representation of allele
specific copy numbers if they differ in copy number states.
The FILTER field has the following additional applied filters.
• binCount—CNV events with a bin count lower than the threshold are filtered.
• lowModelConfidence—A low confidence in the model estimate marks all records as non-PASSING.
The FORMAT fields are described in the header section. The following fields are specific to somatic
CNV:
• AS—Number of allelic read count sites.
• BC—Number of read count bins.
• CN—Estimated total copy number in tumor fraction of sample.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
151
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• CNF—Floating point estimate of tumor copy number.
• CNQ—Exact total copy number Qscore.
• MAF—Maximum a posteriori estimate of minor allele frequency.
• MCN—Estimated minor-haplotype copy number.
• MCNF—Floating point estimate of tumor minor-haplotype copy number.
• MCNQ—Minor copy number Qscore.
• NCN—Normal-sample copy number (present only in germline-aware mode).
• SCND—Difference between CN and GCN (present only in germline-aware mode).
• SD—Best estimate of segment’s bias-corrected read count.
Allele Specific Copy Number Example
By estimating tumor purity, the total tumor copy number can be reported. The BAF estimates, whether
from matched normal SNVs or population SNPs, allows for allele specific copy number calling.
The following table provides examples for a DUP in a reference diploid region.
Total Copy
Number
Minor Copy
Number
ASCN
Scenario
4 2 2+2
4 1 3+1
4 0 4+0
The last entry is a loss of heterozygosity (LOH) case. The total copy number is still considered a DUP, so
this is annotated as GAINLOH to distinguish it from copy neutral LOH CNLOH, which would be 2+0.
Repeat Expansion Detection with Expansion Hunter
Short tandem repeats (STRs) are regions of the genome consisting of repetitions of short DNA
segments called repeat units. STRs can expand to lengths beyond the normal range and cause
mutations called repeat expansions. Repeat expansions are responsible for many diseases, including
Fragile X syndrome, amyotrophic lateral sclerosis, and Huntington's disease.
DRAGEN includes a repeat expansion detection method called ExpansionHunter. ExpansionHunter
performs sequence-graph based realignment of reads that originate inside and around each target
repeat. It then genotypes the length of the repeat in each allele based on these graph alignments.
More information and analysis is available in the following ExpansionHunter papers:
• ExpansionHunter (http://www.genome.org/cgi/doi/10.1101/gr.225672.117)
• Graph ExpansionHunter (https://doi.org/10.1101/572545)
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
152
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
These methods work only for whole human genome samples in PCR-free libraries. Repeats are only
genotyped if the coverage at the locus is at least 10x.
Repeat Expansion Detection Options
To enable DRAGEN repeat expansion detection, the following command-line options are required.
• --repeat-genotype-enable = true
• --repeat-genotype-specs=<path to spec file>
In addition, the sex of the sample should be set using the --sample-sex option.
The following options are optional.
• --repeat-genotype-region-extension-length=<length of region around repeat to examine>
(default 1000bp)
• --repeat-genotype-min-baseq=<Minimum base quality for ‘high confidence’ bases> (default 20)
For more information on the spec file specified by --repeat-genotype-specs option, see Repeat
Expansion Specification Files on page 1.
The main output of repeat expansion detection is a VCF file, containing the variants found via this
analysis.
Repeat Expansion Specification Files
The repeat-specification (also called variant catalog) JSON file defines the repeat regions for
ExpansionHunter to analyze. Default repeat-specification for some pathogenic repeats are in the
/opt/edico/repeat-specs/_directory (based on the reference genome used with DRAGEN).
You can create specification files for new repeat regions by using one of the provided specification files
as a template. See the ExpansionHunter documentation for details on the format.
`--repeat-genotype-specs` is required for ExpansionHunter. If the option is not provided, DRAGEN will
attempt to auto-detect the applicable catalog file from /opt/edico/repeat-specs/ based on the
reference provided.
Repeat Expansion Detection Output Files
VCF Output File
The results of repeat genotyping are output as a separate VCF file, giving the length of each allele at
each callable repeat defined in the repeat-specification catalog file. The name is
<outputPrefix>.repeats.vcf (.gz).
The VCF output file begins with the following fields.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
153
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Field Description
CHROM Chromosome identifier
POS Position of the first base before the repeat region in the reference
ID Always .
REF The reference base at position POS
ALT List of repeat alleles in format <STRn> where n is the number of repeat units
QUAL Always .
FILTER LowDepth filter is applied when the overall locus depth is below 10x or the number of
reads that span one or both breakends is below 5.
Table 4 Core VCFFields
Field Description
SVTYPE Always STR
END Position of the last base of the repeat region in the reference
REF Number of repeat units spanned by the repeat in the
reference
RL Reference length in bp
RU Repeat unit in the reference orientation
REPID Repeat id from the repeat-specification file
Table 5 Additional INFOFields
Field Description
GT Genotype
SO Type of reads that support the allele; can be SPANNING, FLANKING, or INREPEAT
meaning that the reads span, flank, or are fully contained in the repeat
CI Confidence interval called repeat length of each allele
AD_SP Number of spanning reads consistent with the allele
AD_FL Number of flanking reads consistent with the allele
AD_IR Number of in-repeat reads consistent with the allele
Table 6 GENOTYPE (Per Sample) Fields
For example, the following VCF entry describes the state of C9orf72 repeat in a sample with ID
LP6005616-DNA_A03.
QUAL FILTER INFO FORMAT LP6005616-DNA_A03
chr9 27573526 . C <STR2>,<STR349> .
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
154
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
PASS
SVTYPE=STR;END=27573544;REF=3;RL=18;RU=GGCCCC;REPID=ALS
GT:SO:CN:CI:AD_SP:AD_FL:AD_IR
1/2:SPANNING/INREPEAT:2/349:2-2/323-376:19/0:3/6:0/459
In this example, the first allele spans 2 repeat units while the second allele spans 349 repeat units. The
repeat unit is GGCCCC (RU INFO field), so the sequence of the first allele is GGCCCCGGCCCC and the
sequence of the second allele is GGCCCC x 349. The repeat spans three repeat units in the reference
(REF INFO field).
The length of the short allele was estimated from spanning reads (SPANNING) while the length of the
expanded allele was estimated from in-repeat reads (INREPEAT). The confidence interval for the size
of the expanded allele is (323,376). There are 19 spanning and 3 flanking reads consistent with the
repeat allele of size 2 (that is 19 reads fully contain the repeat of size 2 and 2 flanking reads overlap at
most 2 repeat units). Also, there are 6 flanking and 459 in-repeat reads consistent with the repeat
allele of size 349.
Additional Output Files
The sequence-graph alignments of reads in the targeted repeat regions are output in a BAM file. You
can use a specialized GraphAlignmentViewer tool (github.com/Illumina/GraphAlignmentViewer/) to
visualize the alignments. Programs like Integrative Genomics Viewer (IGV) are not designed for
displaying graph-aligned reads and cannot visualize these BAMS.
The BAMs store graph alignments in custom XG tags using the format
<LocusName>,<StartPosition>,<GraphCIGAR>.
• LocusName—A locus identifier that matches the corresponding entry in the repeat expansion
specification file.
• StartPosition—The starting alignment position of a read on the first graph node.
• GraphCIGAR—The alignment of a read against the graph starting from that position. GraphCIGAR
consists of a sequence of graph node identifiers and linear CIGARS describing the alignment of the
read to each node.
Quality scores in the BAM file are binary. High-scoring bases are assigned a score of 40, and low-
scoring bases are assigned a score of 0.
Spinal Muscular Atrophy Calling
Disruption of all copies of the SMN1 gene in an individual causes spinal muscular atrophy (SMA). SMN1
has a very high identity paralog, SMN2, with differs only in approximately 10 SNVs and small indels. One
of these (hg19 chr5:70247773 C->T) affects splicing and largely disrupts the production of functional
SMN protein from SMN2. Standard WGS analysis does not produce complete variant calling results for
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
155
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

SMN due to this high-similarity duplication combined with common copy-number variation. However,
approximately 95% of SMA cases can be detected by determining the absence of the functional C
(SMN1) allele in any copy of SMN.
DRAGEN SMA calling uses sequence-graph realignment to align reads to a single reference
representing SMN1 and SMN2. In addition to the standard diploid genotype call, DRAGEN uses a direct
statistical test to check for presence of any C allele. If no C allele is detected, the sample is called
affected, otherwise unaffected.
SMA calling is only supported for human whole-genome sequencing samples in PCR-free libraries.
Usage
SMA calling is implemented together with repeat expansion detection. For information on graph-
alignment and options, see Repeat Expansion Detection with Expansion Hunter on page 152.
SMA calling is enabled, along with repeat expansion detection, by setting the --repeat-genotype-
enable option to true. To activate SMA calling, the variant specification catalog file must include a
description of the targeted SMN1/2 variant. Example files are available in the /opt/edico/repeat-
specs/experimental folder.
SMN output is included along with any targeted repeats in <outputPrefix>.repeat.vcf. SMNoutput is
represented as a single SNV call at the key (splice-affecting) position in SMN1, with SMA status in
custom fields:
Field Description
VARID SMN marks the SMN call.
GT Genotype call at this position using a normal (diploid) genotype model.
DST SMA status call: + indicates detected, - indicates undetected, ? indicates undetermined.
AD Total read counts supporting the C and T allele.
RPL Log10 Likelihood ratio between the affected and unaffected models. Positive scores are in
favor of unaffected.
Table 7 SMAResult in repeat.vcf Output
CYP2D6 Caller
The CYP2D6 caller is capable of genotyping the CYP2D6 gene from whole-genome sequencing (WGS)
data and is derived from the method implemented in Cyrius¹. Due to high sequence similarity with its
pseudogene paralog CYP2D7 and a wide variety of common structural variants (SVs), a specialized
caller is necessary to resolve variants and identify likely star allele haplotypes.
The CYP2D6 Caller performs the following steps:
1. Determines total CYP2D6 and CYP2D7 copy number from read depth.
2. Determines CYP2D6-derived copy number at CYP2D6/CYP2D7 differentiating sites.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
156
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

3. Detects SV breakpoints by calculating the changes in CYP2D6-derived copy number along the
CYP2D6 gene.
4. Calls small variants in CYP2D6 copies.
5. Identifies star alleles from the detected SV breakpoints and small variants.
6. Identifies the most likely genotype for the called star alleles.
The CYP2D6 Caller requires whole-genome sequencing (WGS) data aligned to a human reference
genome with at least 30x coverage.
Total CYP2D6 and CYP2D7 Copy Number
The first step of CYP2D6 calling is to determine the combined copy number of CYP2D6 and CYP2D7.
Reads aligned to regions in either CYP2D6 or CYP2D7 are counted. The counts in each region are
corrected for GC-bias, and then normalized to a diploid baseline. The GC-bias correction and
normalization factors are determined from read counts in 3000 preselected 2 kb regions across the
genome. The combined CYP2D6 and CYP2D7 copy number is then calculated from the average
sequencing depth across the CYP2D6 and CYP2D7 regions.
Differentiating Sites
The CYP2D6-derived copy number is calculated at 117 predefined differentiating sites across the
CYP2D6 gene. The differentiating sites are selected at positions with sequence differences in CYP2D6
and CYP2D7 where calling the CYP2D6-derived copy number was most likely to be correct based on
sequencing data from the 1000 Genomes Project. The figure below shows the frequency of correctly
calling the CYP2D6-derived copy number as a function of the position having a sequence difference
between CYP2D6 and CYP2D7. The sequence differences showing calling accuracy > 98% are used as
differentiating sites.
For each differentiating site, CYP2D6-specific and CYP2D7-specific alleles are counted in reads
mapping to either CYP2D6 or the homologous region in CYP2D7. The CYP2D6-derived copy number is
then calculated from this two gene-specific allele counts using the total CYP2D6 and CYP2D7 copy
number calculated from the previous step.
Structural Variant Calling
The CYP2D6-derived copy number along the CYP2D6 gene is used to identify known population
structural variants (SVs), including whole gene deletions and duplications as well as certain gene
conversions and gene fusions. The following fusion variants are detected:
Fusion Breakpoint Hybrid Gene Structure Associated Star Alleles
exon 9 2D6-2D7 4.013, 36, 57, 83
exon 9 2D7-2D6 13
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
157
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Fusion Breakpoint Hybrid Gene Structure Associated Star Alleles
intron 4 2D7-2D6 13
intron 1 2D7-2D6 13
intron 1 2D6-2D7 68
In addition to the exon 9 fusion breakpoints, exon 9 can participate in CYP2D7 gene conversion
resulting in an embedded CYP2D7 sequence instead of a true hybrid. The exon 9 gene conversions are
also detected by the structural variant caller. Because only changes in CYP2D6-derived copy number
yield structural variant calls, there might be rare cases where two hybrid copies result in no structural
variant calls. For example, when both *36 and *13 with fusion breakpoint in exon 9 are present.
However, the structural variant caller is capable of detecting multiple copies of the same fusion type
(eg, *36x2) or cases where both an exon 9 gene conversion copy and an exon 9 2D6-2D7 hybrid are
present.
Small Variant Calling
118 small variants that define various star alleles are detected from the read alignments. 96 of these
variants are in unique (nonhomologous) regions of CYP2D6 with high mapping quality. Only reads
mapping to CYP2D6 are used for calling variants in nonhomologous regions. The other 22 variants
occur in homologous regions of CYP2D6 where reads mapping to either CYP2D6 or CYP2D7 are used
for variant calling.
For each variant, reads containing either the variant allele or the nonvariant alleles are counted. A
binomial model that incorporates the sequencing errors is then used to determine the most likely
variant copy number (0 for nonvariant). A strand bias filter is applied for certain noisy variants that tend
to have false positive calls.
In some cases, when the allele counts are noisy or the total CYP2D6 copy number is greater than five,
the most likely variant copy number can be wrong. To handle these cases, the small variant caller also
indicates alternate, less likely variant copy numbers.
Star Allele Identification
The called SVs and small variant genotypes are matched against the definitions of 124 different star
alleles. In some cases, different sets of star alleles might match the called variant genotypes. The
matching sets are emitted. When the small variant caller indicates alternate variant copy numbers with
significant likelihood, then these alternate sets of variant genotypes are also matched to the star allele
definitions. The number of matched star alleles must match the number of CYP2D6-derived gene
copies determined from previous steps. When there are fewer than two CYP2D6-derived gene copies,
then one or more *5 deletion haplotypes are included in the output set of star alleles. If all variant
genotypes cannot be matched to a set of star alleles, the CYP2D6 caller returns a no call during the
genotyping step with filter value No_call.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
158
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Genotyping
Given a possible set of star alleles, the genotyping step attempts to identify the two likely haplotypes
that contain all star alleles in the set. The deletion haplotype\(*5) is considered as a possible haplotype
during this process. The likelihood of any given genotype is determined from a table of population
frequencies determined from the 1000 Genomes Project and the most likely genotype is selected.
When two or more possible genotypes are identified with similar likelihoods, then all genotypes are
emitted. This results in a call with filter value More_than_one_possible_genotype.
CYP2D6 Output File
The CYP2D6 Caller generates a <output-file-prefix>.cyp2d6.tsv file in the output directory.
The output file contains the following tab-delimited fields:
• Sample name.
• One or more semicolon-delimited CYP2D6 genotypes or None for no call.
• The filter status. The value can include: PASS, No_call, or More_than_one_possible_genotype.
Each CYP2D6 genotype contains two haplotypes separated by a slash (eg *1/*2). Each haplotype
consists of one or more star alleles separated by a plus sign (eg *10+*36). When a haplotype contains
more than one copy of the same star allele, that star allele only appears once and is followed by a
multiplication sign, and then the number of copies (eg *1x2 for two copies of *1).
Output File Examples
The following are example output files:
NA18632 *10+*36x2/*52 PASS
HG01190 *4+*68/*5 PASS
NA17244 *2/*4x2+*13+*83;*2/*4+*4.013+*13+*39 More_than_one_possible_
genotype
NA19908 *1/*46;*43/*45 More_than_one_possible_genotype
NA18611 None No_call
Command-line Examples
To enable the CYP2D6 Caller, use --enable-cyp2d6=true. The CYP2D6 Caller is disabled by
default. The CYP2D6 Caller can run directly with the mapper or from prealigned BAM/CRAM input. You
can also enable the CYP2D6 Caller in parallel with other components as part of a germline analysis
workflow.
FASTQ Input
The following is an example command line with FASTQ input:
dragen \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
159
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
-r /staging/human/reference/hg38_alt_aware/DRAGEN/8 \
--fastq-file1 /staging/test/data/NA12878_R1.fastq \
--fastq-file2 /staging/test/data/NA12878_R2.fastq \
--output-directory /staging/test/output \
--output-file-prefix NA12878_dragen \
--RGID DRAGEN_RGID \
--RGSM NA12878 \
--enable-map-align=true \
--enable-cyp2d6=true
Prealigned BAM Input
The following is an example command line with prealigned BAM input:
dragen \
-r /staging/human/reference/hg38_alt_aware/DRAGEN/8 \
--bam-input /staging/test/data/NA12878.bam \
--output-directory /staging/test/output \
--output-file-prefix NA12878_dragen \
--enable-map-align=false \
--enable-cyp2d6=true
Structural Variant Calling
The DRAGEN Structural Variant (SV) Caller integrates and extends Manta structural variant caller
methods to provide structural variant (SV) and indel calls 50 bases or larger. SVs and indels are called
from mapped paired-end sequencing reads. The SV caller is optimized for analysis of germline variation
in small sets of individuals .
The SV caller performs the following actions:
• Discovers, assembles, and scores large-scale SVs, medium-sized indels, and large insertions within
a single efficient workflow.
• Combines paired and split-read evidence during SV discovery and scoring to improve accuracy, but
does not require split-reads or successful breakpoint assemblies to report a variant in cases where
there is strong evidence otherwise.
• Scores known SV deletions and insertions from an input VCF file against one or more input
samples, either as a standalone procedure or together with standard SV discovery.
• Provides scoring models for germline variants in small sets of diploid samples .
All SV and indel inferences are output in VCF 4.1 format.
DRAGEN SV Caller Overview
The DRAGEN SV Caller divides the SV and indel discovery process into the following two primary steps:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
160
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
1. Scans the genome to build various genome-wide data structures, including a breakend association
graph of all SV associated regions. The graph contains edges connecting all regions of the genome
that have a possible breakend association. Edges can connect two different regions of the genome
to represent evidence of a long-range association, or an edge may connect a region to itself to
capture a local indel/small SV association. These associations are more general than a specific SV
hypothesis and many breakend candidates may be found on one edge. Typically only one or two
candidates are found per edge.
2. Analyzes breakend association graph to discover candidate SVs, then scores discovered candidate
SVs and any known SVs from the input. Analysis and scoring is performed as follows.
a. Inference of SV candidates associated with the given graph edge.
b. Attempted assembly of the SVs breakends.
c. Scoring/genotyping and filtration of the SV under various biological models (currently diploid,
germline, and somatic).
d. Output scored SVs to VCF.
DRAGEN SV Caller Capabilities
The Consumable Prefix SV Caller can discover all structural variant types that are identifiable in the
absence of copy number analysis and large-scale de novo assembly. For more information on
detectable types, see Detected Variant Classes on page 162.
For each structural variant and indel, the SV Caller attempts to assemble the breakends to base pair
resolution and report the left-shifted breakend coordinate (per the VCF 4.1 SV reporting guidelines),
together with any breakend homology sequence and/or inserted sequence between the breakends. It
is often the case that the assembly will fail to provide a confident explanation of the data. In this case,
the variant is reported as IMPRECISE, and scored according to the paired-end read evidence only.
You can provide known SVs to be scored as input. This known SV input can be scored either standalone
or together with the standard SV discovery workflow, in which case the known and discovered SVs are
merged.
The sequencing reads provided as input to the SV Caller are expected to be from a paired-end
sequencing assay that results in an "innie" orientation between the two reads of each sequence
fragment, each presenting a read from the outer edge of the fragment insert inward.
The SV Caller is primarily tested for whole-genome and whole-exome (or other targeted enrichment)
sequencing assays on DNA. For these assays the following applications are supported:
• Joint analysis of 10 or fewer diploid individuals
• Subtractive analysis of a matched tumor/normal sample pair
• Analysis of an individual tumor sample
For joint analysis, there is no specific restriction against larger cohorts, but this has not been
extensively tested so there might be stability or call quality issues.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
161
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Tumor samples can be analyzed without a matched normal sample. In this case, no scoring function is
available, but the supporting evidence counts are available and many filters can still be usefully applied.
Detected Variant Classes
The SV Caller can discover all variation classes that can be explained as novel DNA adjacencies in the
genome. Novel DNA adjacencies are classified into the following categories based on the breakend
pattern:
• Deletions
• Insertions. SV insertions can be divided into the following two subclasses depending on if the
inserted sequence can be fully assembled.
– Fully-assembled insertions
– Partially-assembled (ie. inferred) insertions
• Tandem Duplications
• Unclassified breakend pairs corresponding to intra- and inter-chromosomal translocations, or
complex structural variants.
Known Limitations
The SV Caller should not be able to directly discover the following variant types:
• Dispersed duplications.
• Most expansion/contraction variants of a reference tandem repeat.
• Breakends corresponding to small inversions.
– The limiting size is not tested, but in theory detection falls off below ~200 bases. So-called
micro-inversions might be detected indirectly as combined insertion/deletion variants.
• Fully-assembled large insertions.
– The maximum fully-assembled insertion size should correspond to approximately twice the
read-pair fragment size, but power to fully assemble the insertion should fall off to impractical
levels before this size.
– The SV Caller does detect and report very large insertions when the breakend signature of such
an event is found, even though the inserted sequence cannot be fully assembled.
More general repeat-based limitations exist for all variant types:
• Power to assemble variants to breakend resolution falls to zero as breakend repeat length
approaches the read size.
• Power to detect any breakend falls to (nearly) zero as the breakend repeat length approaches the
fragment size.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
162
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
While the SV Caller classifies certain novel DNA-adjacencies into variant classes, it has limited ability to
infer high-level events resulting from complex rearrangements, so certain calls summarized as
deletions, duplications, and insertions
might be better described by looking at the full system of breakends and copy number changes
associated with a given event.
Forced Genotyping Capability
The DRAGEN SV caller is capable of force genotyping a set of SVs input from a VCF file. Forced
genotyping means that the input SVs are scored and emitted in the output of the SV Caller even if the
variant is not supported in the sample data. For example, given a germline analysis, the input variants
are processed and written to the output VCF, even if the variant quality threshold falls below the
normally required for an SV to be emitted.
Forced genotyping typically enables known SVs to be detected at higher recall than standard SV
discovery (particularly for SV discovery on a lower-depth sample). Forced genotyping can also be
useful to assert against the presence of an SV allele. For example, you can use forced genotyping to
distinguish a confident homozygous reference genotype from a lack of sequencing coverage over the
SV locus.
Forced genotyping SVs are processed according to the current SV analysis being run. For example if a
germline analysis is configured by providing one or more normal samples as input, then the input SVs
are scored under a germline model.
Forced genotyping alleles are always emitted in the output and might have modified scoring and
filtering rules applied compared to SVs only discovered from the sample data.
Forced Genotyping Modes
Forced Genotyping can be run in two modes.
• Standalone—Only SVs described in an input VCF are scored and emitted.
• Integrated—The standard SV discovery analysis is run and the results are merged with SVs scored
from the forced genotyping input. The workflow outputs the union of SVs discovered from the
sample data and any additional forced genotyping alleles. The workflow is run whenever the --sv-
discovery option is true.
Forced Genotyping Inputs
You can specify forced genotyping input using the --sv-forcegt-vcf option. The input must be a VCF of
SV alleles. The SV allele types are restricted to insertions and deletions, which are not labeled with the
VCF IMPRECISE flag. The following are the filtering criteria required for the VCF record to be
processed as an input SV allele. Any of the filtration criteria lead to the VCF record being removed from
the set of input SVs for forced genotyping. When a forced genotyping VCF is specified on the command
line, the SV caller reports the total number of SV records used as input SVs and the total number of
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
163
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
records filtered (if any) due to the above criteria.
• Describes an insertion or deletion. Deletions can optionally be described using the symbolic ALT
format (<DEL>) when a value is also provided for INFO/END. Insertions must have an ALT entry
describing the complete insertion sequence.
• Cannot contain the INFO/IMPRECISE flag.
• Cannot contain multiple ALT alleles.
• Has a FILTER value of PASS or .
• All indels are greater than the minimum scored variant size (default is 50).
• Cannot contain a previous SV allele.
• The REF field cannot be empty or unknown (.)
Forced Genotyping Output
Forced genotyping SVs are always output to the standard VCF output of the SV Caller, regardless of
whether the forced genotyping is standalone or integrated with SV calling. When the same SV allele is
independently discovered from the sample data, only the discovered SV appears in the final output.
The discovered SV allele is annotated to indicate the match to a forced genotyping input SV, and the
scoring and filtration rules are changed to match.
VCF output records influenced by forced genotyping have the following associated fields.
• The flag INFO/NotDiscovered is set for any VCF record that was not independently discovered
from the sample data. When forced genotyping is run in standalone, all output records contain the
flag. When integrated with SV calling, the flag can distinguish the SV alleles that would not have
been discovered in a standard SV analysis.
– For these variants only, the usual SV caller ID field generated from the SV Locus graph is not
available, instead the ID is taken from the corresponding user input VCF. The suffix
UserInput${InputVCFRecordNumber} is appended to the ID, separated by an underscore.
• Any output VCF record that corresponds to a forced genotyping input VCF record has the value
INFO/UserInputId=${ID} set to reflect the VCF ID value of the input VCF record. The
corresponding record might have also been discovered independently from the sample data and
might not have the INFO/NotDiscovered flag set.
• Any output VCF record that corresponds to a forced genotyping input VCF record containing forced
genotyping alleles that match exactly to an input SV have the flag INFO/KnownSVScoring. VCF
records with this flag are always emitted in the output of the SV Caller. Several filters, such as
MaxDepth, are not applied.
Input Requirements
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
164
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
When running the SV Caller, the input sequencing reads must be from a standard Illumina paired-end
sequencing assay with an FR read pair orientation, where for each sequence fragment, a read
proceeds from each end of the fragment inwards.
The SV Caller is optimized for paired end libraries where the fragment size is typically larger than the
size of both reads. Overlapping read pairs can be used to discover SVs, but might not always be
handled optimally. For libraries where the typical fragment size is less than the read length, the SV
caller attempts to differentiate reads sequencing into adapter sequence from variant signal. In such
cases, the SV Caller's input quality checks may fail and cause SV analysis to be skipped.
Input Quality Checks
The SV Caller runs quality checks on the input sequencing reads for each sample to make sure that the
input corresponds to a paired read assay with the expected FR orientation, prior to estimating the
fragment size distribution. To check consensus read pair orientation, a subset of high quality read pairs
is sampled. At least 90% of these must have the expected FR orientation for SV analysis to continue,
otherwise the SV caller issues a warning, skips any further analysis, and the resulting output files
display empty results.
The SV Caller can tolerate nonpaired reads in the input, if sufficient paired-end reads exist to estimate
the fragment size distribution. To estimate the fragment size distribution, the SV Caller requires at
least 100 read pairs which meet the quality requirements of the estimation routine. Both reads of the
pair must have a non-zero mapping quality to the same chromosome, are not filtered or part of a split
read mapping, and do not contain indels or soft-clipping. If a sample does not contain a sufficient
number of such read pairs, the SV Caller issues a warning, skips any further analysis, and writes empty
results to its output files.
Read Groups
The SV Caller disregards any read group labels applied to the input sequences. Each input sample is
treated as a separate library with a single fragment size distribution.
File Format
In standalone mode, input sequencing reads must be mapped and provided as input in either BAM or
CRAM format. Each input file must be coordinate sorted and indexed to produce an asamtools/htslib-
style index in a file named to match the input BAM or CRAM file with an additional '.bai', '.crai' or '.csi' file
name extension.
At least one BAM or CRAM file must be provided for the normal or tumor sample. A matched tumor-
normal sample pair can be provided as well. If multiple input files are provided for the normal sample,
each file is treated as a separate sample as part of a joint diploid sample analysis.
In standalone mode, input BAM or CRAM files contain the following limitations:
• Alignments cannot have an unknown read sequence (SEQ="*")
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
165
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Alignments cannot contain the "=" character in the SEQ field.
• Alignments cannot use the sequence match/mismatch ("="/"X"). CIGAR notation RG (read group)
tags in the alignment records are ignored. Each alignment file is treated as representing one
sample.
• Alignments with base call quality values greater than 70 are rejected. These are not supported on
the assumption that this indicates an offset error.
Exome/Targeted Mode
The SV caller can be configured for targeted sequencing inputs, which disables high depth filters.
Exome mode can be directly set to true or false with the command line option --sv-exome. If not
directly set, exome mode defaults to false unless the you run the SV caller in integrated mode and
there is not more than 50 Gb of sequencing input.
Structural Variant Calling Options
The following command line options are supported for the Structural Variant Caller:
• --enable-sv—Enable or disable the structural variant caller. The default is false.
• --sv-call-regions-bed—Specifies a BED file containing the set of regions to call. Optionally, you can
compress the file in gzip or bgzip format.
• --sv-region—Limit the analysis to a specified region of the genome for debugging purposes. This
option can be specified multiple times to build a list of regions. The value must be in the format
“chr:startPos-endPos”.
• --sv-exome—Set to true to configure the variant caller for targeted sequencing inputs, which
includes disabling high depth filters. In integrated mode, the default is to autodetect targeted
sequencing input, and in standalone mode the default is false.
• --sv-output-contigs—Set to true to have assembled contig sequences output in a VCF file. The
default is false.
• --sv-forcegt-vcf—Specify a VCF of structural variants for forced genotyping. The variants are
scored and emitted in the output VCF even if not found in the sample data. The variants are
merged with any additional variants discovered directly from the sample data.
• --sv-discovery—Flag to enable SV discovery. This flag can be set to false only when --sv-forcegt-
vcf is used. When set to false, SV discovery is disabled and only the forced genotyping input
variants are processed. The default is true.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
166
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Modes of Operation
Structural Variant calling can run in the following modes:
• Standalone—Uses mapped BAM/CRAM input files. This mode requires the following options:
– --enable-map-align false
– --enable-sv true
• Integrated—Automatically runs on the output of the DRAGEN mapper/aligner. This mode requires
the following options:
– --enable-map-align true
– --enable-sv true
– --enable-map-align-output true
– --output-format bam
Structural Variant calling can be enabled along with any other caller as well.
The following is an example command line for Integrated mode:
dragen -f \
--ref-dir=<HASH_TABLE> \
--enable-map-align true \
--enable-map-align-output true \
--enable-sv true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX> \
--RGID Illumina_RGID \
--RGSM <sample name> \
-1 <FASTQ1> \
-2 <FASTQ2>
The following is an example command line for Joint Diploid calling in standalone mode:
dragen -f \
--ref-dir <HASH_TABLE> \
--bam-input <BAM1> \
--bam-input <BAM2> \
--bam-input <BAM3> \
--enable-map-align false \
--enable-sv true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX>
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
167
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Structural Variant VCFOutput
The structural variants VCF output file is available in the output directory. The file is named <output-
file-prefix>.sv.vcf.gz. The contents of the file depend on the type of analysis.
For each major analysis category (germline, tumor/normal and tumor-only), the appropriate VCF
output file is output, reflecting variant calls made under the variant calling mode corresponding to the
given analysis type.
The Structural Variant caller produces additional output in the <output-directory>/sv/ directory.
The <output-directory>/sv/results folder contains additional variants and statistics output files.
Additional subdirectories contain logs and intermediate outputs from the variant calling process.
Structural Variant Predictions
In <output-directory>/sv/results/variants, the SV Caller outputs a set of VCF files. Currently,
two VCF files are created for a germline analysis, and an additional somatic VCF is produced for a
tumor/normal subtraction. These files are:
• diploidSV.vcf.gz
SVs and indels scored and genotyped under a diploid model for the set of samples in a joint diploid
sample analysis or for the normal sample in a tumor/normal subtraction analysis. In the case of a
tumor/normal subtraction, the scores in this file do not reflect any information from the tumor
sample.
• somaticSV.vcf.gz
SVs and indels scored under a somatic variant model. This file is only produced if a tumor sample
alignment file is supplied during configuration
• candidateSV.vcf.gz
Unscored SV and indel candidates. Only a minimal amount of supporting evidence is required for a
variant to be entered as a candidate in this file. A variant must be a candidate to be considered for
scoring, therefore a variant does not appear in the other VCF outputs if it is not present in the file.
The file includes indels of size 8 and larger. The smallest indels are provided for workflow flexibility,
but are not scored. Indel scoring starts at size 50.
For tumor-only analysis, the SV Caller produces the following additional output VCF:
• tumorSV.vcf.gz
Subset of the candidateSV.vcf.gz file after removing redundant candidates and small indels less
than the minimum scored variant size (50). The SVs are not scored, but include the following
additional details: (1) paired and split read supporting evidence counts for each allele, (2) a subset
of the filters from the scored tumor-normal model are applied to the single tumor case to improve
precision.
VCF Output
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
168
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
VCF output follows the VCF 4.1 specification for describing structural variants. It uses standard field
names wherever possible. All custom fields are described in the VCF header. The following sections
provide information on the variant representation details and the primary VCF field values.
VCF Sample Names
Sample names output in the VCF output are extracted from each input alignment file from the first read
group (@RG) record found in the header. Any spaces found in the name are replaced with underscores.
If no sample name is found, a default (SAMPLE1, SAMPLE2, etc.) label is used instead.
Small Indel Representation
All variants are reported in the VCF using symbolic alleles unless they are classified as a small indel, in
which case full sequences are provided for the VCF REF and ALT allele fields. A variant is classified as a
small indel if all of the following criteria are met:
• The variant can be entirely expressed as a combination of inserted and deleted sequence.
• The deletion or insertion length is not 1000 or greater.
• The variant breakends and/or the inserted sequence are not imprecise.
When VCF records are output in the small indel format, they also include the CIGAR INFO tag describing
the combined insertion and deletion event.
Insertions with Incomplete Insert Sequence Assembly
Large insertions are reported in some cases even when the insert sequence cannot be fully assembled.
In this case, the SV Caller reports the insertion using the <INS> symbolic allele and includes the special
INFO fields LEFT_SVINSSEQ and RIGHT_SVINSSEQ to describe the assembled left and right ends of
the insert sequence. The following is an example of such a record from the joint diploid analysis of
NA12878, NA12891 and NA12892 mapped to hg19:
chr1 11830208 MantaINS:1577:0:0:0:3:0 T <INS> 999 PASS
END=11830208;SVTYPE=INS;CIPOS=0,12;CIEND=0,12;HOMLEN=12;HOMSEQ=TAAATTTTTCT
T;LEFT_
SVINSSEQ=TAAATTTTTCTTTTTTCTTTTTTTTTTAAATTTATTTTTTTATTGATAATTCTTGGGTGTTTCTC
ACAGAGGGGGATTTGGCAGGGTCACGGGACAACAGTGGAGGGAAGGTCAGCAGACAAACAAGTGAACAAAGGTC
TCTGGTTTTCCCAGGCAGAGGACCCTGCGGCCTTCCGCAGTGTTCGTGTCCCTGATTACCTGAGATTAGGGATT
TGTGATGACTCCCAACGAGCATGCTGCCTTCAAGCATCTGTTCAACAAAGCACATCTTGCACTGCCCTTAATTC
ATTTAACCCCGAGTGGACACAGCACATGTTTCAAAGAG;RIGHT_
SVINSSEQ=GGGGCAGAGGCGCTCCCCACATCTCAGATGATGGGCGGCCAGGCAGAGACGCTCCTCACTTCCTA
GATGTGATGGCGGCTGGGAAGAGGCGCTCCTCACTTCCTAGATGGGACGGCGGCCGGGCGGAGACGCTCCTCAC
TTTCCAGACTGGGCAGCCAGGCAGAGGGGCTCCTCACATCCCAGACGATGGGCGGCCAGGCAGAGACACTCCCC
ACTTCCCAGACGGGGTGGCGGCCGGGCAGAGGCTGCAATCTCGGCACTTTGGGAGGCCAAGGCAGGCGGCTGCT
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
169
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
CCTTGCCCTCGGGCCCCGCGGGGCCCGTCCGCTCCTCCAGCCGCTGCCTCC GT:FT:GQ:PL:PR:SR
0/1:PASS:999:999,0,999:22,24:22,32 0/1:PASS:999:999,0,999:18,25:24,20
0/0:PASS:230:0,180,999:39,0:34,0
Normalizing Small Tandem Duplications
Tandem duplications may also be represented as insertions. This representation creates ambiguity in
how such variants are represented in the VCF output, particularly for small tandem duplications, and
can lead to complications such as unrecognized call duplication.
To better normalize the SV caller output such that the same variant type is not represented in two
different VCF formats, small tandem duplications (<1000 bases) are converted to insertions in the VCF
output. Insertions converted from such tandem duplications have a formatting similar to incomplete
insertions, using the symbolic allele <INS> for the ALT field. An example of such an insertion is:
chr2 2520057 MantaDUP:TANDEM:53645:0:1:0:0:0 T <INS> 813 PASS
END=2520057;SVTYPE=INS;SVLEN=52;DUPSVLEN=52 GT:FT:GQ:PL:PR:SR
0/1:PASS:393:863,0,390:25,0:19,25
Converted insertions include copies of certain output fields as they would have appeared in a tandem
duplication record, such as INFO/DUPSVINSSEQ providing a copy of the breakpoint insertion value
computed for the duplication. In the context of a duplication such a value would normally be written to
INFO/SVINSSEQ. An example of a converted insertion with such a value is:
chr2 2645730 MantaDUP:TANDEM:53649:0:1:0:0:0 C <INS> 367 PASS
END=2645730;SVTYPE=INS;SVLEN=97;DUPSVLEN=86;DUPSVINSLEN=11;DUPSVINSSEQ=CTC
ACCTTCAT GT:FT:GQ:PL:PR:SR 0/1:PASS:367:417,0,386:19,0:20,15
For more information about copied INFO fields, see VCF INFO Fields on page 171.
Inversions
Inversions are reported as a set of breakends. For example, given a simple reciprocal inversion, four
breakends are reported, sharing the same EVENT INFO tag. The following is an example breakend
records representing a simple reciprocal inversion:
chr1 17124941 MantaBND:1445:0:1:1:3:0:0 T [chr1:234919886[T 999 PASS
SVTYPE=BND;MATEID=MantaBND:1445:0:1:1:3:0:1;CIPOS=0,1;HOMLEN=1;
HOMSEQ=T;INV5;EVENT=MantaBND:1445:0:1:0:0:0:0;JUNCTION_QUAL=254;BND_
DEPTH=107;
MATE_BND_DEPTH=100 GT:FT:GQ:PL:PR:SR 0/1:PASS:999:999,0,999:65,8:15,51
chr1 17124948 MantaBND:1445:0:1:0:0:0:0 T T]chr1:234919824] 999 PASS
SVTYPE=BND;MATEID=MantaBND:1445:0:1:0:0:0:1;INV3;EVENT=MantaBND:1445:0:1:0
:0:0:0;
JUNCTION_QUAL=999;BND_DEPTH=109;MATE_BND_DEPTH=83 GT:FT:GQ:PL:PR:SR
0/1:PASS:999:999,0,999:60,2:0,46
chr1 234919824 MantaBND:1445:0:1:0:0:0:1 G G]chr1:17124948] 999 PASS
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
170
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

SVTYPE=BND;MATEID=MantaBND:1445:0:1:0:0:0:0;INV3;EVENT=MantaBND:1445:0:1:0
:0:0:0;
JUNCTION_QUAL=999;BND_DEPTH=83;MATE_BND_DEPTH=109 GT:FT:GQ:PL:PR:SR
0/1:PASS:999:999,0,999:60,2:0,46
chr1 234919885 MantaBND:1445:0:1:1:3:0:1 A [chr1:17124942[A 999 PASS
SVTYPE=BND;MATEID=MantaBND:1445:0:1:1:3:0:0;CIPOS=0,1;HOMLEN=1;
HOMSEQ=A;INV5;EVENT=MantaBND:1445:0:1:0:0:0:0;JUNCTION_QUAL=254;BND_
DEPTH=100;
MATE_BND_DEPTH=107 GT:FT:GQ:PL:PR:SR 0/1:PASS:999:999,0,999:65,8:15,51
VCF INFO Fields
ID Description
IMPRECISE Flag indicating that the structural variation is imprecise, ie, the exact
breakpoint location is not found
SVTYPE Type of structural variant
SVLEN Difference in length between REF and ALT alleles
END End position of the variant described in this record
CIPOS Confidence interval around POS
CIEND Confidence interval around END
CIGAR CIGAR alignment for each alternate indel allele
MATEID ID of mate breakend
EVENT ID of event associated to breakend
HOMLEN Length of base pair identical homology at event breakpoints
HOMSEQ Sequence of base pair identical homology at event breakpoints
SVINSLEN Length of insertion
SVINSSEQ Sequence of insertion
LEFT_SVINSSEQ Known left side of insertion for an insertion of unknown length
RIGHT_SVINSSEQ Known right side of insertion for an insertion of unknown length
PAIR_COUNT Read pairs supporting this variant where both reads are confidently
mapped
BND_PAIR_COUNT Confidently mapped reads supporting this variant at this breakend
(mapping may not be confident at remote breakend)
UPSTREAM_PAIR_
COUNT
Confidently mapped reads supporting this variant at the upstream
breakend (mapping may not be confident at downstream breakend)
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
171
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

ID Description
DOWNSTREAM_
PAIR_COUNT
Confidently mapped reads supporting this variant at this downstream
breakend (mapping may not be confident at upstream breakend)
BND_DEPTH Read depth at local translocation breakend
MATE_BND_DEPTH Read depth at remote translocation mate breakend
JUNCTION_QUAL If the SV junction is part of an EVENT (ie, a multi-adjacency variant), this
field provides the QUAL value for the adjacency in question only
SOMATIC Flag indicating a somatic variant
SOMATICSCORE Somatic variant quality score
JUNCTION_
SOMATICSCORE
If the SV junction is part of an EVENT (ie, a multi-adjacency variant), this
field provides the SOMATICSCORE value for the adjacency in question
only
CONTIG Assembled contig sequence, if the variant is not imprecise
(with--outputContig)
DUPSVLEN Length of duplicated reference sequence
DUPHOMLEN Length of base pair identical homology at event breakpoints excluding
duplicated reference sequence
DUPHOMSEQ Sequence of base pair identical homology at event breakpoints excluding
duplicated reference sequence
DUPSVINSLEN Length of inserted sequence after duplicated reference sequence
DUPSVINSSEQ Inserted sequence after duplicated reference sequence
NotDiscovered Variant candidate specified by the user and not discovered from input
sequencing data
UserInputId Variant ID from user input VCF
KnownSVScoring Variant is associated with a user specified input variant, therefore
scoring and filtration criteria are relaxed under a stronger prior
assumption of truth
VCF FORMAT Fields
ID Description
GT Genotype
FT Sample filter, 'PASS' indicates that all filters have passed for this sample
GQ Genotype Quality
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
172
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

ID Description
PL Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF
specification
PR Number of spanning read pairs which strongly (Q30) support the REF or ALT alleles
SR Number of split-reads which strongly (Q30) support the REF or ALT alleles
VCF FILTER Fields
Germline
The following table lists the VCF FILTER fields applied to germline VCF output.
ID Level Description
MinQUAL Record QUAL score is less than 20 (not applied to records with KnownSVScoring
flag)
MinGQ Sample GQ score is less than 15 (filter applied at sample level; not applied to
records with KnownSVScoring flag)
Ploidy Record For DEL & DUP variants, the genotypes of overlapping variants (with
similar size) are inconsistent with diploid expectation (not applied to
records with KnownSVScoring flag)
MaxDepth Record Depth is greater than 3x the median chromosome depth near one or
both variant breakends (not applied to records with KnownSVScoring
flag)
MaxMQ0Frac Record For a small variant (<1000 bases), the fraction of reads in all samples
with MAPQ0 around either breakend exceeds 0.4 (not applied to records
with KnownSVScoring flag)
NoPairSupport Record For variants significantly larger than the paired read fragment size, no
paired reads support the alternate allele in any sample (not applied to
records with KnownSVScoring flag)
SampleFT Record No sample passes all the sample-level filters
HomRef Sample Homozygous reference call
Tumor-Normal Somatic
The following table lists the VCF FILTER fields applied to tumor-normal somatic VCF output.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
173
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

ID Level Description
MinSomaticScore Record SOMATICSCORE is less than 30
MaxDepth Record Normal sample site depth is greater than 3x the median chromosome
depth near one or both variant breakends (not applied to records with
KnownSVScoring flag)
MaxMQ0Frac Record For a small variant (< 1000 bases) in the normal sample, the fraction of
reads with MAPQ0 around either breakend exceeds 0.4 (not applied to
records with KnownSVScoring flag)
Tumor-Only
The following table lists the VCF FILTER fields applied to tumor-only VCF output.
ID Level Description
MaxDepth Record Depth is greater than 3x the median chromosome depth near one or both
variant breakends (not applied to records with KnownSVScoring flag)
MaxMQ0Frac Record For a small variant (<1000 bases), the fraction of reads in all samples with
MAPQ0 around either breakend exceeds 0.4 (not applied to records with
KnownSVScoring flag)
Interpretation of VCF Filters
There are two levels of filters: record level (FILTER) and sample level (FORMAT/FT).
Most record-level filters are independent of sample-level filters. However, in a germline analysis, if
none of the samples pass all sample-level filters, the SampleFT filter record-level filter is applied.
Interpretation of INFO/EVENT Field
Some structural variants reported in the VCF, such as translocations, represent a single novel
sequence junction in the sample. The INFO/EVENT field indicates that two or more such junctions are
hypothesized to occur together as part of a single variant event. All individual variant records belonging
to the same event share the same INFO/EVENT string. Note that although such an inference could be
applied after SV calling by analyzing the relative distance and orientation of the called variant
breakpoints, the SV Caller incorporates this event mechanism into the calling process to increase
sensitivity towards such larger-scale events. Given that at least one junction in the event has already
passed standard variant candidacy thresholds, sensitivity is improved by lowering the evidence
thresholds for additional junctions which occur in a pattern consistent with a multijunction event (such
as a reciprocal translocation pair).
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
174
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Although this mechanism could generalize to events including an arbitrary number of junctions, it is
currently limited to two. Thus, at present it is most useful for identifying and improving sensitivity
towards reciprocal translocation pairs.
VCF ID Field
The VCF ID, or identifier, field can be used for annotation, or in the case of BND (breakend) records for
translocations, the ID value is used to link breakend mates or partners. The following is an example of a
VCF ID field from the SV caller.
MantaINS:1577:0:0:0:3:0
The value provided in the IDfield reflects the SV association graph edge(s) from which the SV or indel
was discovered. The value is unique within any single VCF output file produced by the SV Caller, and is
used to link associated breakend records using the standard VCF MATEID key.
Convert SV VCF to BEDPE Format
It can sometimes be convenient to express structural variants in BEDPE format. For such applications
we recommend the script vcfToBedpe available from:
https://github.com/ctsa/svtools
This repository is forked from @hall-lab with modifications to support VCF 4.1 SV format.
BEDPE format greatly reduces structural variant information compared to the SV Caller VCF output. In
particular, breakend orientation, breakend homology, and insertion sequence are lost, in addition to the
ability to define fields for locus and sample specific information. For this reason, Illumina only
recommends BEDPE as a temporary output for applications that require it.
Statistics Output File
Statistics are provided in the files in <output-directory>/sv/results/stats.
• alignmentStatsSummary.txt
Fragment length quantiles for each input alignment file.
• svLocusGraphStats.tsv
Statistics and runtime information pertaining to the SV locus graph.
• svCandidateGenerationStats.tsv
Statistics and runtime information pertaining to the SV candidate generation.
• svCandidateGenerationStats.xml
XML data backing the svCandidateGenerationStats.tsv report.
• diploidSV.sv_metrics.csv
The number of passing SV calls under a diploid model. This file is only produced in the germline
analysis, or tumor/normal analysis.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
175
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• somaticSV.sv_metrics.csv
The number of passing SV calls under a somatic variant model. This file is only produced in the
tumor/normal analysis.
• tumorSV.sv_metrics.csv
The number of passing SV calls in the tumor-only analysis. This file is only produced in the tumor-
only analysis.
Structural Variant De Novo Quality Scoring
De novo quality scoring can be enabled for structural variant joint diploid calling, by setting --sv-
denovo-scoring to true and supplying a pedigree file. This adds FORMAT/DQand FORMAT/DN fields to
the output VCF file to represent a De Novo Quality Score and an associated De Novo call.
The following example shows a command line for enabling the de novo quality scoring for a joint diploid
run.
dragen -f
--ref-dir <HASH_TABLE> \
--bam-input <BAM1> \
--bam-input <BAM2> \
--bam-input <BAM3> \
--enable-map align=false \
--enable-sv=true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX> \
--sv-denovo-scoring true \
--RGID DRAGEN_RGID \
--RGSM <sample name>
--pedigree-file <PED_FILE>
Consumable Prefix can also be run on an existing Structural Variant output VCF containing multiple
samples (ie, a Trio with a Proband and Parents) to generate a modified VCF file that contains
FORMAT/DQ and FORMAT/DN fields (the original file is not changed).
The following example shows a command line for deriving the de novo quality score from an existing SV
trio.
dragen -f \
--variant <TRIO_VCF_FILE> \
--pedigree-file <PED_FILE> \
--enable-map-align false \
--sv-denovo-scoring true \
--output-directory <OUT_DIR> \
--output-file-prefix <PREFIX>
The DQ field is defined as follows:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
176
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

##FORMAT=<ID=DQ,Number=1,Type=Float,Description="Denovo quality">
The DQfield represents a score of the posterior probability of the variant being denovo in the proband.
If it can be calculated, the score in Phred scale is added to the proband, while the other samples are
marked with a period ( . ) to indicate missing.
For example, DQ scores of 13 and 20 would correspond to a posterior probability of a de novo variant of
0.95 and 0.99, respectively.
The DN Field is defines as follows:
##FORMAT=<ID=DN,Number=1,Type=String,Description="Possible values are
'DeNovo' or 'LowDQ'. Threshold for a passing de novo call is DQ >= 20">
DRAGEN compares valid (> 0) DQ scores with a threshold with default score of 20. A score greater than
or equal to the threshold results in DN field of the sample set to DeNovo, while a score below the
threshold is declared to be LowDQ. If there is not a valid DQ score (ie, DQ equals “0” or “.”,) the DN field
is set to “.”
The threshold can be changed by using the --sv-denovo-threshold command line option. For example,
if the threshold needs to be reduced to 10, add --sv-denovo-threshold 10 to the DRAGEN command
line.
The inputs to this function are the VCF file and the Pedigree file that specifies which sample in the trio is
the proband, mother, or father. In the scenario where there are multiple trios specified in the pedigree
file (eg, multi-generation pedigree), DRAGEN automatically detects the trios and assesses the DeNovo
variants on the proband sample of each trio.
Ploidy Calling
Ploidy Estimator
The Ploidy Estimator runs by default. The Ploidy Estimator uses reads from the mapper/aligner to
calculate the sequencing depth of coverage for each autosome and allosome in the human genome.
The sex karyotype of the sample is then estimated using the ratios of the median sex chromosome
coverages to the median autosomal coverage. The sex karyotype is estimated based on the range the
ratios fall in. If the ratios are outside all expected ranges ,then the Ploidy Estimator does not determine
a sex karyotype.
Sex
Karyotype
X Ratio Min
X
Ratio
Max
Y
Ratio
Min
Y
Ratio
Max
XX 0.75 1.25 0.00 0.25
XY 0.25 0.75 0.25 0.75
XXY 0.75 1.25 0.25 0.75
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
177
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Sex
Karyotype
X Ratio Min
X
Ratio
Max
Y
Ratio
Min
Y
Ratio
Max
XYY 0.25 0.75 0.75 1.25
X0 0.25 0.75 0.00 0.25
XXXY 1.25 1.75 0.25 0.75
XXX 1.25 1.75 0.00 0.25
Ploidy estimation can fail if the type of input sequencing data cannot be determined or if there is not
sufficient sequencing coverage in the autosomes. When ploidy estimation fails the estimated median
coverage values will be zero.
When both tumor and matched normal reads are provided as input, the Ploidy Estimator only estimates
sequencing coverage and sex karyotype for the matched normal sample and ignores the tumor reads.
If only tumor reads are provided as input, the Ploidy Estimator estimates sequencing coverage and sex
karyotype for the tumor sample.
Ploidy Estimator Reference Sex Karyotype
The estimated sex karyotype (if determined), is made available to downstream components (eg variant
callers). For components that support using the estimated sex karyotype, refer to the corresponding
section for more information. To overwrite the sex karyotype used by downstream components, use
the --sample-sex command line option. If the value specified is male, then the sex karyotype is XY. If
the value specified is female, then the sex karyotype is XX. The sex karyotype, whether provided via
the command line or estimated, is converted to a reference sex karyotype for use as a baseline in
variant calling.
The following table provides a summary of reference sex karyotypes and variant callers
Sex Karyotype
CNV
Caller
ExpansionHunter
Ploidy
Caller
Small Variant
Caller
SV
Caller
XX XX XX XX XXYY XXYY
XY XY XY XY XY XXYY
XXY XY XX XY XXYY XXYY
XYY XY XX XY XXYY XXYY
X0 XX XX XX XXYY XXYY
XXXY XY XX XY XXYY XXYY
XXX XX XX XX XXYY XXYY
Undetermined XX XX XX XXYY XXYY
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
178
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• The Copy Number Variant Caller utilizes its own estimation module that is largely consistent with
the Ploidy Estimator. Any differences can be attributed to the read counting implementation.
• The Small Variant Caller marks chrY calls with the PloidyConflict filter when the sex karyotype is XX.
• The Structural Variant Caller always treats the sex chromosomes as diploid and does not take into
account the sex karyotype.
Ploidy Estimator Output Metrics
The Ploidy Estimator results, including each normalized per-contig median coverage, is reported in the
<output-file-prefix>.ploidy_estimation_metrics.csv file and in standard output.
The following is an example of the results.
PLOIDY ESTIMATION Autosomal median coverage 44.79
PLOIDY ESTIMATION X median coverage 42.47
PLOIDY ESTIMATION Y median coverage 20.82
PLOIDY ESTIMATION 1 median / Autosomal median 0.95
PLOIDY ESTIMATION 2 median / Autosomal median 1.05
PLOIDY ESTIMATION 3 median / Autosomal median 1.01
PLOIDY ESTIMATION 4 median / Autosomal median 0.99
...
PLOIDY ESTIMATION 22 median / Autosomal median 0.99
PLOIDY ESTIMATION X median / Autosomal median 0.95
PLOIDY ESTIMATION Y median / Autosomal median 0.46
PLOIDY ESTIMATION Ploidy estimation XXY
Ploidy Caller
The Ploidy Caller uses the per contig median coverage values from the Ploidy Estimator to detect
aneuploidy and chromosomal mosaicism in a human germline sample from whole genome sequencing
data.
The Ploidy Caller runs by default except in the following circumstances:
• The Ploidy Estimator cannot determine if the input data is from whole genome sequencing. For
example, data from exome or targeted sequencing.
• The reference genome does not contain the expected 22 autosomes and two allosomes for
humans.
• There is no germline sample. For example, tumor-only analysis.
Calling Model
Chromosomal mosaicism is detected when there is a significant shift in median coverage of a
chromosome compared to the overall autosomal median coverage.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
179
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

The following table displays some examples of expected shifts in coverage for a give aneuploidy and
mosaic fraction.
Neutral Copy Number Variant Copy Number Mosaic Fraction Expected Coverage Shift
2 1 10 % -5%
2 1 5 % -2.5%
2 3 5 % +2.5%
2 3 10 % +5%
The Ploidy Caller models coverage as a normal distribution for both the null (neutral) and the alternative
(mosaic) hypotheses. The two normal distributions have equal mean at the median autosomal
coverage for the sample, but the variance of the alternative normal distribution is greater than that of
the null normal distribution. The baseline variance of the two models at 30x coverage was determined
empirically from a cohort of ~2500 WGS samples. The actual variance used for the two models is
calculated from the baseline variance at 30x coverage, adjusting for the median autosomal coverage of
the sample. Below are the likelihood distributions for the null and alternative hypotheses for a sample
with 35x median autosomal coverage.
Figure 12 Null and Alternative Hypotheses Likelihood Distributions
After apply an empirically estimated prior for chromosomal mosaicism, the Ploidy Caller generates
ploidy calls according to the posterior probability of null and alternative hypotheses as shown below for
a sample with 35x median autosomal sequencing coverage.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
180
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Figure 13 Null and Alternative Hypotheses Posterior Probability
A 35x median autosomal coverage, the threshold for deciding between a neutral (REF) and an
alternative (DEL or DUP) call is roughly at +/- 5% shift in coverage for an autosome. A 100x median
autosomal coverage, the threshold is at roughly +/0 3% shift in coverage for an autosome. A Q20
threshold is used to filter low quality calls.
Ploidy Caller Reference Sex Karyotype
In addition to detecting aneuploidy and chromosomal mosaicism in autosomes where the expected
reference ploidy is two, the Ploidy Caller can also detect these variants in allosomes.
The reference sex karyotype used for making calls on the allosomes is determined from the sex
karyotype of the sample either provided on the command line using --sample-sex or from the Ploidy
Estimator. If the sex karyotype of the sample is not provided on the command line and not determined
by the Ploidy Estimator, then the sex karyotype is assumed to be XX. Whenever the sex karyotype
contains at least one Y chromosome, the reference sex karyotpye is XY. If the sex karyotype does not
contain at least one Y chromosome, then the sex karyotype is XX.
The following table displays each of the possible sex karyotypes for a sample. If the Y chromosome
reference ploidy is zero, then ploidy calling is not performed on the Y chromosome.
Sex Karyotype X Reference Ploidy Y Reference Ploidy
XX 2 0
XY 1 1
XXY 1 1
XYY 1 1
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
181
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Sex Karyotype X Reference Ploidy Y Reference Ploidy
X0 2 0
XXXY 1 1
XXX 2 0
Ploidy Caller Output File
The Ploidy Caller generates a <output-file-prefix>.ploidy.vcf.gz output file in the output directory. The
output file follows the VCF 4.2 Specification. A single record is reported for each reference autosome
and allosome, except for the Y chromosome if the reference sex karotype is XX. Calls are not made for
other sequences in the reference genome, such as mitochondrial DNA, unlocalized or unplaced
sequences, alternate contigs, decoy contigs, or the Epstein-Barr virus sequence.
The following information is provided in the VCF file.
• Meta information—The VCF output file contains common meta-information such as
DRAGENVersion and DRAGEN CommandLine, as well as Ploidy Caller specific information. The VCF
header contains the meta-information for median autosome depth of coverage, the provided sex
karyotype if available, the estimated sex karyotype from the Ploidy Estimator if available, and the
reference sex karyotype. The following is an example of the header lines:
##autosomeDepthOfCoverage=36.635
##providedSexKaryotype=XY
##estimatedSexKaryotype=X0
##referenceSexKaryotype=XY
• FILTER fields—The VCF output file includes the LowQual filter, which filters results with quality
score below 20.
• INFO Fields—The VCF output INFO fields include the following:
– END—End position of the variant described in this record.
– SVTYPE—Type of structural variant.
• Format fields—The VCF output file includes the following format fields. There is no GT FORMAT
field. A variant call in the VCF displays either <DUP> or <DEL> in the ALT column. A non-variant call
displays . in the ALT column. If using the output file for downstream use, a GT field can be added for
variant calls using ./1 for a diploid contig and 1 for a haploid contig. For non-variant calls, use 0/0
for diploid and 0 for haploid.
– DC—Depth of coverage.
– NDC—Normalized depth of coverage.
The following is an example output file.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
182
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
##fileformat=VCFv4.2
...
##autosomeDepthOfCoverage=36.635
##providedSexKaryotype=XY
##estimatedSexKaryotype=X0
##referenceSexKaryotype=XY
##INFO=<ID=END,Number=1,Type=Integer,Description="End position of the
variant described in this record">
##INFO=<ID=SVTYPE,Number=1,Type=String,Description="Type of structural
variant">
##ALT=<ID=DEL,Description="Deletion relative to the reference">
##ALT=<ID=DUP,Description="Region of elevated copy number relative to the
reference">
##FILTER=<ID=LowQual,Description="QUAL below 20">
##FORMAT=<ID=DC,Number=1,Type=Float,Description="Depth of coverage">
##FORMAT=<ID=NDC,Number=1,Type=Float,Description="Normalized depth of
coverage">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT MySampleName
chr1 1 . N . 31.1252 PASS END=248956422 DC:NDC 36.836:1.00549
chr2 1 . N . 31.451 PASS END=242193529 DC:NDC 36.668:1.0009
...
chr21 1 . N . 31.4499 PASS END=46709983 DC:NDC 36.6:0.999045
chr22 1 . N . 28.8148 PASS END=50818468 DC:NDC 37.2:1.01542
chrX 1 . N . 29.7892 PASS END=156040895 DC:NDC 18:0.982667
chrY 1 . N <DEL> 150 PASS END=57227415;SVTYPE=DEL DC:NDC 5.7:0.311178
```
Cell Line Artifacts
Samples derived from cell lines frequently have coverage artifacts that might result in variant ploidy
calls on some chromosomes. Chromosomes 17, 19, and 22 are the most common for the cell line
coverage artifacts. When performing accuracy assessments of ploidy calls on cell line samples, filter
out chromosomes with known cell line artifacts.
QC Metrics and Coverage/Callability Reports
DRAGEN generates pipeline-specific metrics coverage reports during each run. There are four different
groups of metrics that are generated at different stages of the pipeline:
• Mapping and Aligning metrics
• VCF metrics
• Duration (or run time) metrics
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
183
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• Coverage (or enrichment) metrics and reports
The mapping/aligning metrics, VCF metrics, Duration metrics, and a subset of available coverage
reports are autogenerated and do not require any activation or specific commands. Additional
coverage metrics can be enabled, and additional coverage regions can be specified.
Metric calculation is performed during analysis so that it does not impact the DRAGEN run time.
Figure 14 Generation of Metrics and Reports
QC Metrics Output Format
The QC metrics are printed to the standard out humanand CSV files are written to the run output
directory.
• <output prefix>.mapping_metrics.csv
• <output prefix>.vc_metrics.csv
• <output prefix>.time_metrics.csv
• <output prefix>.<coverage region prefix>_coverage_metrics.csv
• <output prefix>.<other coverage reports>.csv
Section RG/Sample Metric Count/Ration/Time Percentage/Seconds
MAPPING/ALIGNING
SUMMARY
Total input
reads
816360354
MAPPING/ALIGNING
SUMMARY
Number of
duplicate
reads
(marked
not
removed)
15779031 1.93
...
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
184
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Section RG/Sample Metric Count/Ration/Time Percentage/Seconds
MAPPING/ALIGNING
PER RG
RGID_1 Total reads
in RG
816360354 100
MAPPING/ALIGNING
PER RG
RGID_1 Number of
duplicate
reads
(marked)
15779031 1.93
...
VARIANT CALLER
SUMMARY
Number of
samples
1
VARIANT CALLER
SUMMARY
Reads
Processed
738031938
...
VARIANT CALLER
PREFILTER
SAMPLE_1 Total 4918287 100
VARIANT CALLER
PREFILTER
SAMPLE_1 Biallelic 4856654 98.75
...
RUN TIME Time
loading
reference
00:18.6 18.65
RUN TIME Time
aligning
reads
19:24.4 1164.42
Mapping and Aligning Metrics
Mapping and aligning metricslike the metrics computed are available on an aggregate level (over all
input data), and on a per read group level. Unless explicitly stated, the metrics units are in reads (ie, not
in terms of pairs or alignments).
• Total input reads—Total number of reads in the input FASTQ files.
• Number of duplicate marked reads—Reads marked as duplicates as a result of the --enable-
duplicate-marking option being used.
• Number of duplicate marked and mate reads removed—Reads marked as duplicates, along with
any mate reads, that are removed when the --remove-duplicates option is used.
• Number of unique reads—Total number of reads minus the duplicate marked reads.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
185
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Reads with mate sequenced—Number of reads with a mate.
• Reads without mate sequenced—Total number of reads minus number of reads with mate
sequenced.
• QC-failed reads—Reads not passing platform/ vendor quality checks (SAM flag 0x200).
• Mapped reads—Total number of mapped reads minus number of unmapped reads
• Number of unique and mapped reads—Number of mapped reads minus number of duplicate
marked reads.
• Unmapped reads—Total number of reads that could not be mapped.
• Singleton reads—Number of reads where the read could be mapped, but the paired mate could not
be read.
• Paired reads—Count of reads in which both reads in the pair are mapped.
• Properly paired reads—Both reads in the pair are mapped and fall within an acceptable range from
each other based on the estimated insert length distribution.
• Not properly paired reads (discordant)—The number of paired reads minus the number of
properly paired reads.
• Paired reads mapped to different chromosomes—The number of reads with a mate, where the
mate was mapped to a different chromosome
• Paired reads mapped to different chromosomes (MAPQ >= 10)—The number of reads with a
MAPQ>10 and with a mate, where the mate was mapped to a different chromosome
• Reads with indel R1—The percentage of R1 reads containing at least 1 indel.
• Reads with indel R2—The percentage of R2 reads containing at least 1 indel.
• Soft-clipped bases R1—The percentage of bases in R1 reads that are soft clipped.
• Soft-clipped bases R2—The percentage of bases in R2 reads that are soft clipped.
• Mismatched bases R1—The number of mismatched bases on R1, which is the sum of SNP count
and indel lengths. It does not count anything within soft clipping, or RNA introns. It also does not
count a mismatch if either the reference base or read base is N.
• Mismatched bases R2—The number of mismatched bases on R2, which is the sum of SNP count
and indel lengths. It does not count anything within soft clipping, or RNA introns. It also does not
count a mismatch if either the reference base or read base is N.
• Mismatched bases R1 (excluding indels)—The number of mismatched bases on R1. The indels
lengths are ignored. It does not count anything within soft clipping, or RNA introns. It also does not
count a mismatch if either the reference base or read base is N.
• Mismatched bases R2 (excluding indels)—The number of mismatched bases on R2. The indels
lengths are ignored. It does not count anything within soft clipping, or RNA introns. It also does not
count a mismatch if either the reference base or read base is N.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
186
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Q30 Bases—The total number of bases with a BQ ≥ 30.
• Q30 Bases R1—The total number of bases on R1 with a BQ ≥ 30.
• Q30 Bases R2—The total number of bases on R2 with a BQ >= 30.
• Q30 Bases (excluding dups & clipped bases)—The number of bases on non-duplicate, and non-
clipped bases with a BQ ≥ 30.
• Histogram of reads map qualities
– Reads with MAPQ [40:inf)
– Reads with MAPQ [30:40)
– Reads with MAPQ [20:30)
– Reads with MAPQ [10:20)
– Reads with MAPQ [0:10)
• Total alignments—Total number of loci reads aligned to with > 0 quality.
• Secondary alignments—Number of secondary alignment loci.
• Supplementary (chimeric) alignments—A chimeric read is split over multiple loci (possibly due to
structural variants). One alignment is referred to as the representative alignment, the other are
supplementary.
• Estimated read length—Total number of input bases divided by the number of reads.
• Histogram—See Histogram Coverage Report on page 196.
• PCT of bases aligned that fell inside the interval region—Number of bases inside the interval
region and the target region divided by the total number of bases aligned.
• Estimated sample contamination—The estimated fraction of reads in a sample that may be from
another human source.
The prediction accuracy of variant calling is affected by cross-sample contamination. Even small
levels of contamination can lead to many FP calls, especially in pipelines where the aim is to detect
variants with low allele frequencies.
The DRAGEN cross-sample contamination module uses a probabilistic mixture model to estimate
the fraction of reads in a sample that may be from another human source. This sample
contamination fraction is estimated as the parameter value in the mixture model that maximizes
the likelihood of the observed reads at multiple pileup locations. The mixture model accounts for
the population allele frequencies and the inferred sample genotypes.
To enable this metric in germline mode, you must provide the file path on the command line to a
VCF that includes marker sites (RSIDs) with population allele frequencies.
--qc-cross-cont-vcf /opt/edico/config/sample_cross_contamination_
resource_hg19/GRCh37/GRCh38.vcf
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
187
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
In somatic mode the contamination algorithm first tries to avoid biases that could be introduced by
CNV or LoH. The algorithm then estimates nucleotide noise from the sample to adjust for FFPE
samples.
To enable somatic contamination detection, use the following setting.
--qc-somatic-contam-vcf /opt/edico/config/somatic_sample_cross_
contamination_resource_hg19/GRCh37/GRCh38.vcf.gz
The VCF resource files that are included with DRAGEN can be reconstructed from the Ensembl
database. The VCFs included in the DRAGEN config folder contain ~5000 marker locations where
the population AFs are close to 0.5. The files are reference specific (hg19/GRCh37/hg38). DRAGEN
will abort if an incompatible resource and reference file is used (eg, CRCh37 resource file and hg19
reference).
The following shows example output for a sample with 1.1% contamination. This value is provided
as a fraction, so a value of 0.011 the same as 1.1% estimated contamination.
MAPPING/ALIGNING SUMMARY Estimated sample contamination 0.011
Somatic Mode Mapping and Aligning Metrics
In somatic tumor-normal mode, the mapping and aligning metrics are generated separately for the
tumor and normal samples, with each line beginning with TUMOR or NORMAL to indicate the sample.
The metrics for the tumor sample are output first, followed by the metrics for the normal sample.
Metrics per read group are also separated into tumor and normal read groups.
In somatic tumor-only mode, the mapping and aligning metrics follow the same conventions as
germline mode, without metrics labeled as TUMOR or Normal.
Variant Calling Metrics
The generated variant calling metrics are similar to the metrics computed by RTG vcfstats. Metrics are
reported for each sample in multi sample VCF and gVCF files. Based on the run case, metrics are
reported either as standard VARIANT CALLER or JOINT CALLER. Metrics are reported both for the raw
(PREFILTER) and hard filtered (POSTFILTER) VCF file.
PON (Panel of Normals) and COSMIC filtered variants are counted as PASS variants in the POSTFILTER
VCF metrics. These PASS variants can cause higher than expected variant counts in the POSTFILTER
VCF metrics.
• Number of samples—Number of samples in the population/ joint VCF.
• Reads Processed—The number of reads used for variant calling, excluding any duplicate marked
reads and reads falling outside of the target region.
• Total—The total number of variants (SNPs + MNPs + indels).
• Biallelic—Number of sites in a genome that contains two observed alleles, counting the reference
as one, and therefore allowing for one variant allele.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
188
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Multiallelic—Number of sites in the VCF that contain three or more observed alleles. The
reference is counted as one, therefore allowing for two or more variant alleles.
• SNPs—A variant is counted as an SNP when the reference, allele 1, and allele2 are all length 1.
• Insertions (Hom)—Number of variants that contains homozygous insertions
• Insertions (Het)—Number of variants where both alleles are insertions, but not homozygous
• Deletions (Het) —Number of variants that contains homozygous deletions
• INDELS (Het)—Number of variants where genotypes are either [insertion+deletion],
[insertion+SNP] or [deletion+SNP].
• De Novo SNPs—De novo marked SNPs, with DQ > 0.05. Sett the --qc-snp-denovo-quality-
threshold option to the required threshold. The default is 0.05.
• De Novo INDELs—De novo marked indels with DQ values > 0.02. This DQ threshold can be
specified by Setting the --qc-indel-denovo-quality-threshold option to the required DQthreshold.
The default is 0.02.
• De Novo MNPs—Same as the option for SNPs. Set the --qc-snp-denovo-quality-threshold to the
required threshold. The default is 0.05.
• (Chr X SNPs)/(Chr Y SNPs) ratio in the genome (or the target region)—Number of SNPs in
chromosome X (or in the intersection of chromosome X with the target region) divided by the
number of SNPs in chromosome Y (or in the intersection of chromosome Y with the target region).
If there was no alignment to either chromosome X or chromosome Y, this metric shows as NA.
• SNP Transitions—An interchange of two purines (A<->G) or two pyrimidines (C<->T).
• SNP Transversions—An interchange of purine and pyrimidine bases Ti/Tv ratio: ratio of transitions
to transitions.
• Heterozygous—Number of heterozygous variants.
• Homozygous—Number of homozygous variants.
• Het/Hom ratio—Heterozygous/ homozygous ratio.
• In dbSNP—Number of variants detected that are present in the dbSNP reference file. If no dbSNP
file is provided via the --bsnp option, then both the In dbSNP and Novel metrics show as NA.
• Novel—Total number of variants minus number of variants in dbSNP.
• Percent Callability—Available only in germline mode with gVCF output. The percentage of non-N
reference positions having a PASSing genotype call. Multi-allelic variants are not counted.
Deletions are counted for all the deleted reference positions only for homozygous calls. Only
autosomes and chromosomes X, Y, and M are considered.
• Percent Autosome Callability—Only autosomes are considered.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
189
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Percent QC Region Callability in Region i (i is equivalent to regions 1,2, or 3)—Available if
callability for custom regions is requested via the --qc-coverage-region-i option and the callability
output is specified with --qc-coverage-reports-i. All contigs are considered.
Duration Metrics
The duration metrics section includes a breakdown of the run duration for each process. For example,
the following metrics are generated for the mapper and variant caller pipeline:
• Time loading reference
• Time aligning reads
• Time sorting and marking duplicates
• Time DRAGStr calibration
• Time partial reconfiguration
• Time variant calling
• Total runtime
Callability Report
DRAGEN automatically generates a callability report as part of variant caller metrics when running the
germline small variant caller with gVCF output mode.DRAGEN appends the percent callability for each
region included in the BED file specified by the test and generates a full-callability-report in both BED
and CSV formats. Callability is defined as the fraction of non-N reference positions having a PASSing
genotype call. The following criteria are used to calculate callability metrics:
• Callability is calculated over the gVCF.
• Multi-allelic variants are not counted.
• Decoy contigs are ignored.
• Unplaced and unlocalized contigs are ignored.
• N positions are considered non-callable.
• For regions where no variant calling was performed, callability is 0.
• A homozygous deletion counts as a PASSing genotype call for all the reference positions spanned
by the deletion.
If the --vc-target-bed option is specified, the output is a target_bed_callability.bed file that
contains the overall and autosome callability over the input target bed region. The padding size
specified by the --vc-target-bed-padding option is used and overlapping regions are merged.
Callability can also be output with the custom region coverage/callability reports.
Coverage/Callability Reports Over Custom Regions
DRAGEN generates the following coverage reports:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
190
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• A set of default reports for either the whole genome, or, if the --vc-target-bed option is specified,
for the target region.
• Optionally, additional reports for up to three regions of interest (coverage regions).
For each specified region, DRAGEN generates the default reports, and any optional report
requested for the region.
To generate coverage region reports, use the –qc-coverage-region-i option, where i is 1, 2, or 3.
• Each –qc-coverage-region-i option requires a bed file argument.
• Regions in each bed file can be optionally padded using the --qc-coverage-region-padding-i option,
which defaults to 0.
• A set of default reports is generated for each region.
• Optionally, additional reports can be specified for each region by using the –qc-coverage-reports-i
option.
The following example shows the options required to generate coverage reports.
$dragen … \
--qc-coverage-region-1 <bed file 1> \
--qc-coverage-reports-1 full_res \
--qc-coverage-region-2 <bed file 2> \
--qc-coverage-region-3 <bed file 3> \
--qc-coverage-reports-3 full_res cov_report
Counting Reads and Bases
All default and optional coverage reports listed in Table 8 and Table 9 use the following default rules for
counting reads and bases:
• Duplicate reads are ignored.
• Soft and/or hard-clipped bases are ignored.
• Reads with MAPQ=0 are ignored.
• Overlapping mates are double-counted.
Nondefault settings:
The reports are available with or without running either the mapper and aligner, or the variant caller.
However, the --enable-sort options must be set to true (the default is true).
By default overlapping mates are double counted. Set --qc-coverage-ignore-overlaps=true to resolve
all of the alignments for each fragment and avoid double-counting any overlapping bases. This might
result in marginally longer run times. This option also requires setting --enable-map-align=true. --gc-
coverage-ignore-overlaps is a global setting and updates all qc-coverage-reports.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
191
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
By default soft-clipped bases are not counted towards coverage. Set --qc-coverage-count-soft-
clipped-bases=true to include soft-clipped bases in the coverage calculations. --qc-coverage-count-
soft-clipped-bases is a global setting and updates all qc-coverage reports.
Any combination of the optional reports can be requested for each region. If multiple report types are
selected per region, they should be space-separated.
It is possible to override the min MAPQ and min BQ to apply for a given region using the qc-coverage-
filters.
A coverage filter is enabled by using one of the --qc-coverage-filters-i options (where i is 1, 2, or 3), in
combination with the associated --qc-coverage-region-i option:
• --qc-coverage-region-i=<targetedregions.bed>
• --qc-coverage-filters-i <filters string>
For example, the following options are used to enable 1 bp resolution coverage output with filtering:
--qc-coverage-region-1 <targetedregions.bed>
--qc-coverage-filters-1 'mapq<10,bq<30'
--qc-coverage-reports-1 full_res
• The argument syntax is mapq<value,bq<value, which means that reads that have a mapping
quality less than the specified value are not counted, and/or bases with a base call quality below
the specified value.
• Valid filter arguments are mapq and bq only. Either, or both, can be specified.
• Only one operator < is supported. <=, >, >=, = are not supported.
• When filtering is enabled for a targeted region, DRAGEN outputs the filtered report files for this
region. Unfiltered report files are not output for the targeted filtered region.
Callability for the Custom Regions
If the --qc-coverage-region-i option is used with --qc-coverage-reports-i (where i is 1, 2, or 3),
callability can be added as a report type for that region. The output is a qc-coverage-region-i_
callability.bed file. For each specified qc-coverage-region-i file, the average callability is
reported in the variant calling metrics file. The padding size specified by the --qc-coverage-region-
padding-i is used and overlapping regions are merged.
The optional min MAPQ and min BQ filters only influence read and base counting and do not influence
the callability reports.
Contig lengths and region of interest lengths (used as denominators) do not include regions with N in
the FASTA.
Available Report Types
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
192
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

File Name Description
_coverage_
metrics.csv
Coverage Metrics Report on page
193
_fine_hist.csv Fine Histogram Coverage Report
on page 196
_hist.csv Histogram Coverage Report on
page 196
_overall_mean_
cov.csv
Overall Mean Coverage Report on
page 196
_contig_mean_
cov.csv
Per Contig Mean Coverage Report
on page 196
_ploidy.csv Predicted Ploidy Report
Table 8 Default Report
File Name Description
_full_res.bed Full Res Report on page 196
_cov_report.bed Coverage Report on page 197
_callability.bed Callability Report on page 190
Table 9 Optional Reports
Coverage Metrics Report
The coverage metrics report outputs a _coverage_metrics.csv file, which provides metrics over a
region, where the region can be the genome, a target region, or a QC coverage region. The first column
of the output file contains the section name COVERAGE SUMMARY and the second column is empty for
all metrics.
The following criteria are used when calculating coverage:
• Duplicate reads and clipped bases are ignored.
• Only reads with MAPQ > min MAPQ and bases with BQ > min BQ are considered.
The following table lists the calculated metrics:
Metric Description
Aligned bases in
region
Number of uniquely mapped bases to region and the percentage
relative to the number of uniquely mapped bases to the genome.
Average alignment
coverage over region
Number of uniquely mapped bases to region divided by the number of
sites in region.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
193
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Metric Description
Uniformity of
coverage (PCT >
0.2*mean) over
region
Percentage of sites with coverage greater than 20% of the mean
coverage in region.
PCT of region with
coverage [ix, inf)
Percentage of sites in region with at least ix coverage, where i can
equal 100, 50, 20, 15, 10, 3, 1, or 0.
PCT of region with
coverage [ix, jx)
Percentage of sites in region with at least ix but less than jx coverage,
where (i, j) can equal (50, 100), (20, 50), (15, 20), (10, 15), (3, 10), (1, 3),
or (0, 1).
Average
chromosome X
coverage over region
Total number of bases that aligned to the intersection of chromosome
X with region divided by the total number of loci in the intersection of
chromosome X with region. If there is no chromosome X in the
reference genome or the region does not intersect chromosome X, this
metric shows as NA.
Average
chromosome Y
coverage over region
Total number of bases that aligned to the intersection of chromosome
Y with region divided by the total number of loci in the intersection of
chromosome Y with region. If there is no chromosome Y in the
reference genome or the region does not intersect chromosome Y, this
metric shows as NA.
XAvgCov/YAvgCov
ratio over
genome/target
region
Average chromosome X alignment coverage in region divided by the
average chromosome Y alignment coverage in region. If there is no
chromosome X or chromosome Y in the reference genome or the
region does not intersect chromosome X or Y, this metric shows as NA.
Average
mitochondrial
coverage over region
Total number of bases that aligned to the intersection of the
mitochondrial chromosome with region divided by the total number of
loci in the intersection of the mitochondrial chromosome with region. If
there is no mitochondrial chromosome in the reference genome or the
region does not intersect mitochondrial chromosome, this metric
shows as NA.
Average autosomal
coverage over region
Total number of bases that aligned to the autosomal loci in region
divided by the total number of loci in the autosomal loci in region. If
there is no autosome in the reference genome, or the region does not
intersect autosomes, this metric shows as NA.
Median autosomal
coverage over
region—
Median alignment coverage over the autosomal loci in region. If there
is no autosome in the reference genome or the region does not
intersect autosomes, this metric shows as NA.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
194
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Metric Description
Mean/Median
autosomal coverage
ratio over region
Mean autosomal coverage in region divided by the median autosomal
coverage in region. If there is no autosome in the reference genome or
the region does not intersect autosomes, this metric shows as NA.
Aligned reads in
region—
Number of uniquely mapped reads to region and percentage relative to
the number of uniquely mapped reads to the genome. When region is
the target BED, this metric is equivalent to and replaces Capture
Specificity based on target region.
The following is an example of the contents of the _coverage_metrics.csv file:
COVERAGE SUMMARY,,Aligned bases,148169295474
COVERAGE SUMMARY,,Aligned bases in genome,148169295474,100.00
COVERAGE SUMMARY,,Average alignment coverage over genome,46.08
COVERAGE SUMMARY,,Uniformity of coverage (PCT > 0.2*mean) over genome,91.01
COVERAGE SUMMARY,,PCT of genome with coverage [100x: inf),0.25
COVERAGE SUMMARY,,PCT of genome with coverage [50x: inf),50.01
COVERAGE SUMMARY,,PCT of genome with coverage [20x: inf),89.46
COVERAGE SUMMARY,,PCT of genome with coverage [15x: inf),90.51
COVERAGE SUMMARY,,PCT of genome with coverage [10x: inf),91.01
COVERAGE SUMMARY,,PCT of genome with coverage [ 3x: inf),91.69
COVERAGE SUMMARY,,PCT of genome with coverage [ 1x: inf),92.10
COVERAGE SUMMARY,,PCT of genome with coverage [ 0x: inf),100.00
COVERAGE SUMMARY,,PCT of genome with coverage [50x:100x),49.76
COVERAGE SUMMARY,,PCT of genome with coverage [20x: 50x),39.45
COVERAGE SUMMARY,,PCT of genome with coverage [15x: 20x),1.04
COVERAGE SUMMARY,,PCT of genome with coverage [10x: 15x),0.51
COVERAGE SUMMARY,,PCT of genome with coverage [ 3x: 10x),0.67
COVERAGE SUMMARY,,PCT of genome with coverage [ 1x: 3x),0.42
COVERAGE SUMMARY,,PCT of genome with coverage [ 0x: 1x),7.90
COVERAGE SUMMARY,,Average chr X coverage over genome,24.70
COVERAGE SUMMARY,,Average chr Y coverage over genome,20.96
COVERAGE SUMMARY,,Average mitochondrial coverage over genome,20682.19
COVERAGE SUMMARY,,Average autosomal coverage over genome,47.81
COVERAGE SUMMARY,,Median autosomal coverage over genome,48.62
COVERAGE SUMMARY,,Mean/Median autosomal coverage ratio over genome,0.98
COVERAGE SUMMARY,,XAvgCov/YAvgCov ratio over genome,1.18
COVERAGE SUMMARY,,XAvgCov/AutosomalAvgCov ratio over genome,0.52
COVERAGE SUMMARY,,YAvgCov/AutosomalAvgCov ratio over genome,0.44
COVERAGE SUMMARY,,Aligned reads,1477121058
COVERAGE SUMMARY,,Aligned reads in genome,1477121058,100.00
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
195
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Fine Histogram Coverage Report
The fine histogram report outputs a _fine_hist.csv file, which contains two columns: Depth and
Overall. The value in the Depth column ranges from 0 to 1000+ and the Overall column indicates the
number of loci covered at the corresponding depth.
Histogram Coverage Report
The histogram report outputs a _hist.csv file, which provides the following information:
• Percentage of bases in the coverage bed/target bed/wgs region that fall within a certain range of
coverage.
• Duplicate reads are ignored if DRAGEN is run with --enable-duplicate-marking true.
The following ranges are used:
"[100x:inf)", "[1x:3x)", "[0x:1x)"
Overall Mean Coverage Report
The Overall Mean Coverage report generates an _overall_mean_cov.csv file, which contains the
average alignment coverage over the coverage bed/target bed/wgs, as applicable.
The following is an example of the contents of the _overall_mean_cov.csv file:
Average alignment coverage over target_bed,80.69
Per Contig Mean Coverage Report
The Contig Mean Coverage report generates a _contig_mean_cov.csv file, which contains the
estimated coverage for all contigs, and an autosomal estimated coverage. The file includes the
following three columns:
Column 1 Column 2 Column 3
Contig name Number of bases aligned to that
contig, which excludes bases
from duplicate marked reads,
reads with MAPQ=0, and clipped
bases.
The estimated covered,
calculated as follows.
Number of bases aligned to the
contig (ie, Col2) divided by length
of the contig or (if a target bed is
used) the total length of the
target region spanning that
contig.
Full Res Report
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
196
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

The Full Res Report outputs a _full_res.bed file. This file is tab-delimited file and has as its first three
columns the standard BED fields, and as its fourth column the depth. Each record in the file is for a
given interval that has a constant depth. If the depth changes, then a new record is written to the file.
Alignments that have a mapping quality value of 0, duplicate reads, and clipped bases are not counted
towards the depth.
Only base positions that fall under the user-specified coverage-region bed regions are present in the _
full_res.bed output file.
The _full_res.bed file structure is similar to the output file of bedtools genomecov -bg. The contents are
identical if the bedtools command line is executed after filtering out alignments with mapping quality
value of 0, and possibly filtering by a target BED (if specified).
The following is an example of the contents of the _full_res.bed file:
chr1 121483984 121483985 10
chr1 121483985 121483986 9
chr1 121483986 121483989 8
chr1 121483989 121483991 7
chr1 121483991 121483992 6
chr1 121483992 121483993 4
chr1 121483993 121483994 2
chr1 121483994 121484039 1
chr1 121484039 121484043 2
chr1 121484043 121484048 3
chr1 121484048 121484050 7
chr1 121484050 121484051 11
chr1 121484051 121484052 17
chr1 121484052 121484053 149
chr1 121484053 121484054 323
chr1 121484054 121484055 2414
Coverage Report
The cov_report report generates a _cov_report.bed file in a tab-delimited formatfull-coverage-report
file in both BED and CSV formats. The first three columns are standard BED fields. The remaining
column fields are statistics calculated over the interval region specified on the same record line.
The following table lists the appended columns.
Column Description
total_cvg The total coverage value.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
197
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Column Description
mean_cvg The mean coverage value.
Q1_cv The lower quartile (25th percentile) coverage value.
median_cvg The median coverage value.
Q3_cvg The upper quartile (75th percentile) coverage value.
min_cvg The minimum coverage value.
max_cvg The maximum coverage value.
pct_above_X Indicates the percentage of bases over the specified interval region that
had a depth coverage greater than X.
By default, if an interval has a total coverage of 0, then the record is written to the output file. To filter
out intervals with zero coverage, set vc-emit-zero-coverage-intervals to false in the configuration file.
The following is an example of the contents of the _cov_report.bed file:
chrom start end total_cvg mean_cvg Q1_cvg median_cvg Q3_cvg min_cvg max_cvg pct_above_5
…
chr5 34190121 34191570 76636 52.89 44.00 54.00 60.00 32 76 100.00
…
chr5 34191751 34192380 39994 63.58 57.00 61.00 69.00 50 85 100.00
…
chr5 34192440 34192642 10074 49.87 47.00 49.00 51.00 44 62 100.00
…
chr9 66456991 66457682 31926 46.20 39.00 45.00 52.00 27 65 100.00
…
chr9 68426500 68426601 4870 48.22 42.00 48.00 54.00 39 58 100.00
…
chr17 41465818 41466180 24470 67.60 4.00 66.00 124.00 2 153 66.30
…
chr20 29652081 29652203 5738 47.03 40.00 49.00 52.00 34 58 100.00
…
chr21 9826182 9826283 4160 41.19 23.00 52.00 58.00 5 60 99.01
…
Coverage/Callability Reports Use Cases and Expected Output
The following table shows which outputs are generated when default options (--vc-target-bed) versus
optional coverage region options (--coverage-region) are used.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
198
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

--vc-
target-bed
specified?
Y/N
--qc- coverage-region-i
(i equal to 1, 2, or 3)
specified? Y/N
Expected Output Files
N N wgs_coverage_metrics.csv
wgs_fine_hist.csv
wgs_hist.csv
wgs_overall_mean_cov.csv
wgs_contig_mean_cov.csv
N Y wgs_coverage_metrics.csv
wgs_fine_hist.csv
wgs_hist.csv
wgs_overall_mean_cov.csv
wgs_contig_mean_cov.csv
For each coverage region specified by the user:
qc-coverage-region-i_coverage_metrics.csv
qc-coverage-region-i_fine_hist.csv
qc-coverage-region-i_hist.csv
qc-coverage-region-i_overall_mean_cov.csv
qc-coverage-region-i_contig_mean_cov.csv
qc-coverage-region-i_full_res.bed if full_res report
type is requested for qc-coverage-region-i
qc-coverage-region-i_cov_report.bed if cov_report
report type is requested for qc-coverage-region-i
qc-coverage-region-i_callability.bed if GVCF mode is
enabled and the callability or exome-callability report
type is requested
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
199
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

--vc-
target-bed
specified?
Y/N
--qc- coverage-region-i
(i equal to 1, 2, or 3)
specified? Y/N
Expected Output Files
Y N wgs_coverage_metrics.csv
wgs_fine_hist.csv
wgs_hist.csv
wgs_overall_mean_cov.csv
wgs_contig_mean_cov.csv
target_bed_coverage_metrics.csv
target_bed_fine_hist.csvtarget_bed_hist.csv
target_bed_overall_mean_cov.csv
target_bed_contig_mean_cov.csv
target_bed_callability.bed if GVCF mode is enabled
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
200
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

--vc-
target-bed
specified?
Y/N
--qc- coverage-region-i
(i equal to 1, 2, or 3)
specified? Y/N
Expected Output Files
Y Y wgs_coverage_metrics.csv
wgs_fine_hist.csv
wgs_hist.csv
wgs_overall_mean_cov.csv
wgs_contig_mean_cov.csv
target_bed_coverage_metrics.csv
target_bed_fine_hist.csv
target_bed_hist.csv
target_bed_overall_mean_cov.csv
target_bed_contig_mean_cov.csv
target_bed_callability.bed if GVCF mode is enabled
For each coverage region specified by the user:
qc-coverage-region-i _coverage_metrics.csv
qc-coverage-region-i_fine_hist.csv
qc-coverage-region-i_hist.csv
qc-coverage-region-i_overall_mean_cov.csv
qc-coverage-region-i_contig_mean_cov.csv
qc-coverage-region-i_full_res.bed if full_res report
type is requested for qc-coverage-region-i
qc-coverage-region-i_cov_report.bed if cov_report
report type is requested for qc-coverage-region-i
qc-coverage-region-i_callability.bed if GVCF mode is
enabled and the callability or exome-callability report
type is requested
BigWig Compression of Coverage Metrics
When --enable-metrics-compression is set to true, the 1 bp resolution coverage metrics output bed file
(_full_res.bed) are compressed to bigwig format.
GC Bias Report
The GC bias report provides information on GC content and the associated read coverage across a
genome. DRAGEN GC bias metric is modeled after the Picard implementation and adapted to
preexisting internal measures. The DRAGEN GC bias correction module attempts to correct these
biases following the target count stage. For more information, see GC Bias Correction on page 127
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
201
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The GC bias metric is computed as follows.
1. Calculates GC content using a 100 bp wide, per-base rolling window over all chromosomes in the
reference genome, excluding any decoys and alternate contigs. Windows containing more than
four masked (N) bases in the reference are discarded.
2. Calculates the average coverage for each window, excluding any non-PF, duplicate, secondary,
and supplementary reads.
3. Calculates the average global coverage across the whole genome.
4. Groups valid windows based on the percentage of GC content, both at individual percentages and
five 20% ranges as summary.
5. Calculates the normalized coverage for each group by dividing the average coverage for the bin by
the global average coverage across the genome. Values below 1.0 indicate a lower than expected
coverage at the given GC percent or range. Coverages significantly deviating from 1.0 at greater
GC values are an expected result.
6. Calculates dropout metrics as the sum of all positive values of (percentage of windows at GC X-
percentage aligned reads at GC X) for each GC ≤ 50% and > 50% for AT and GC dropout.
By default, the GC bias metric report is not calculated. To enable GC Bias calculations, enter the --gc-
metrics-enable command line option. The following is an example command:
$dragen -b <BAM file> -r <reference genome> --gc-metrics-enable=true
The GC metrics report generates a gc_metrics.csv file. The file is structured as follows.
GC BIAS DETAILS,,Windows at GC [0-100],<number of windows>,<fraction of all
windows>
GC BIAS DETAILS,,Normalized coverage at GC [0-100],<average coverage of all
windows at given GC divided by average coverage of whole genome>
GC METRICS SUMMARY,,Window size,<window size in base, typically 100>
GC METRICS SUMMARY,,Number of valid windows,<total number of windows used
in calculations>
GC METRICS SUMMARY,,Number of discarded windows,<total number windows
discarded due to more than 4 masked bases>
GC METRICS SUMMARY,,Average reference GC,<average GC content over all valid
windows>
GC METRICS SUMMARY,,Mean global coverage,<average genome coverage over all
valid windows>
GC METRICS SUMMARY,,Normalized coverage at GCs <GC range>,<average coverage
of all windows at given GC range divided by average coverage of whole
genome>
GC METRICS SUMMARY,,AT Dropout,<Calculated AT dropout value>
GC METRICS SUMMARY,,GC Dropout,<Calculated GC dropout value>
The GC bias report also includes the following command line options, but they are not recommended.
• --gc-metrics-window-size—Overrides the default rolling window size of 100 bp.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
202
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• --gc-metrics-num-bins—Overrides the number of summary bins.
Somatic Metrics Report
In somatic tumor-normal mode, DRAGEN generates separate reports for the tumor and normal
samples. Each report is labeled according to the sample type. Tumor sample reports include tumor at
the end of the file name, and normal sample reports include normal at the end of the file name. To
include both tumor and normal sample results in one file, set the --vc-enable-separate-t-n-metrics
option to false. DRAGEN then reports on the aggregate of both samples instead.
DRAGEN generates the following reports:
• coverage_metrics
• contig_mean_cov
• fine_hist
• hist
• overall_mean_cov
• ploidy
• cov_report (if requested)
• full_res (if requested)
• gc_metrics (if requested)
If a target bed is included in the run or the option --qc-region-coverage-region-i (where i is 1, 2, or 3) is
included, then the corresponding coverage reports are also split into tumor and normal files.
In somatic tumor-only mode, the reports are identical to those in germline mode and do not include
tumor or normal in the file names.
Somatic Callable Regions Report
In somatic mode, DRAGEN automatically generates a somatic callable regions report as a bed file. The
somatic callable regions report includes all regions with tumor coverage at least as high as the tumor
threshold and (if applicable) normal coverage at least as high as the normal threshold. If only the tumor
sample is provided, then the report includes all regions with tumor coverage at least as high as the
tumor threshold. Each line in the bed output file is formatted as follows.
chromosome region_start region_end
You can specify the threshold values using the --vc-callability-tumor-thresh or --vc-callability-normal-
thresh options. The default value for the tumor threshold is 15. The default value for the normal
threshold is 5. For more information on each option, see Somatic Mode Options on page 96.
If the target bed or the --qc-region-coverage-region-i(where i is 1, 2, or 3) option is included in the run.
The DRAGEN then generates corresponding somatic callable regions bed files in addition to the whole
genome somatic callable region bed file.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
203
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Virtual Long Read Detection
DRAGEN Virtual Long Read Detection (VLRD) is an alternate and more accuratevariant caller focused
on processing homologous/similar regions of the genome. A conventional variant caller relies on the
mapper/aligner to determine which reads likely originated from a given location. It also detects the
underlying sequence at that location independently of other regions not immediately adjacent to
it.Conventional variant calling works well when the region of interest does not resemble any other
region of the genome over the span of a single read (or a pair of reads for paired-end sequencing).
However, a significant fraction of the human genome does not meet this criterion.Many regions of the
genome have near-identical copies elsewhere, and as a result, the true source location of a read might
be subject to considerable uncertainty. If a group of reads is mapped with low confidence, a typical
variant caller might ignore the reads, even though they contain useful information. If a read is
mismapped (ie, the primary alignment is not the true source of the read), it can result in detection
errors. Short-read sequencing technologies are especially susceptible to these problems. Long-read
sequencing can mitigate these problems, but it typically has much higher cost and/or higher error rates,
or other shortcomings.
DRAGEN VLRD attempts to tackle the complexities presented by the genome's redundancy from a
perspective driven by the short-read data. Instead of considering each region in isolation, VLRD
considers all locations from which a group of reads may have originated and attempts to detect the
underlying sequences jointly using all available information.
Running DRAGEN VLRD
Like the DRAGEN variant caller, VLRD takes either FASTQ or sorted BAM files as input and produces an
output VCF file.VLRD supports processing a set of only two homologous regions.
VLRD is not enabled by default. To run VLRD, set the --enable-vlrd option to true. The following is an
example DRAGEN command to run VLRD.
dragen \
-r <REF> \
-1 <FQ1> \
-2 <FQ2> \
--RGID <RG> --RGSM <SM> \
--output-dir <OUTPUT> \
--output-file-prefix <PREFIX> \
--enable-map-align true \
--enable-sort=true \
--enable-duplicate-marking true \
--enable-vlrd true
--vc-target-bed similar_regions.bed
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
204
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
VLRD Updated Map/Align Output
DRAGEN VLRD can output a remapped BAM/SAM file in addition to the regular DRAGEN Map/Align
output. To enable output of a remapped BAM/SAM file, set the --enable-vlrd-map-align-output option
to true. The default for this option is false.
The additional map/align output by VLRD only contains reads mapped to the regions that were
processed by VLRD.
VLRD considers the information available from all the homologous regions jointly to update the read
alignments. The updated VLRD map/align output is primarily useful for pile-up analysis involving
homologous regions.
VLRD Settings
The following options are specific to VLRD in the DRAGEN host software.
• --enable-vlrd
If set to true, VLRD is enabled for the DRAGEN pipeline.
• --vc-target-bed
Specifies the input bed file. DRAGENrequires an input target bed file specifying the homologous
regions to be processed by VLRD. The input bed file id formatted to correctly process the
homologous regions. The maximum region span processed by VLRD is 900 bp.
For example:
chr1 161497562 161498362 0 0
chr1 161579204 161580004 0 0
chr1 21750837 217516371 0
chr1 21809355 218101551 1
• The first three columns are like traditional bed files: column 1 is chromosome description,
column 2 is region start, and column 3 is region end.
• Column 4 is homologous region Group ID. This groups regions that are homologous to each
other.
• Rows 1 and 2 have the same value in column 4 indicating that these should be processed as a
set of homologous regions, independent of the next group in row 3 and 4. If not set correctly,
software might group regions that are not homologous to each other, leading to incorrect
variant calls.
• Column 5 indicates whether a region is reverse complemented with respect to the other
homologous region. A value of 1 denotes that the region is reverse complemented with respect
to other regions in the same group.
• Row 4, column 5 is set to 1. This indicates that the region is homologous to the region in row 3
only if it is reverse complemented.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
205
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The DRAGEN installation package contains two VRLD bed files for hg19 and hs37d5 reference
genomes under /opt/edico/examples/VLRD. You can use these bed files as is for running VLRD,
or as an example to generate a custom bed file.
• --enable-vlrd-map-align-output
If set to true, VLRD outputs a remapped BAM/SAM file that only contains reads mapped to the
regions that were processed by VLRD.
Unique Molecular Identifiers
DRAGEN can process data from whole genome and hybrid-capture assays with unique molecular
identifiers (UMI). UMIs are molecular tags added to DNA fragments before amplification to determine
the original input DNA molecule of the amplified fragments. UMIs help reduce errors and biases
introduced by DNA damage such as deamination before library prep, PCR error, or sequencing errors.
To use the UMI Pipeline, the input reads files must be from a paired-end run. Input can be pairs of
FASTQ files or aligned/unaligned BAM input.
DRAGEN supports the following UMI types:
• Dual, nonrandom UMIs, such as TruSight Oncology (TSO) UMI Reagents or IDT xGen Prism.
• Dual, random UMIs, such as Agilent SureSelect XT HS2 molecular barcodes (MBC) or IDT xGen
Duplex Seq Adapters.
• Single-ended, random UMIs, such as Agilent SureSelect XT HS molecular barcodes (MBC) or IDT
xGen dual index UMI Adapters.
DRAGEN uses the UMI sequence to group the read pairs by their original input fragment and generates
a consensus read pair for each such group, or family. The consensus reduces error rates to detect rare
and low frequency somatic variants in DNA samples with high accuracy. DRAGEN generates a
consensus as follows.
1. Aligns reads.
2. Groups reads into groups with matching UMI and pair alignments. These groups are referred to as
families.
3. Generates a single consensus read pair for each read family.
These generated reads have higher quality scores than the input reads and reflect the increased
confidence gained by combining multiple observations into each base call.
UMI workflow is only compatible with small variant calling and SV in DRAGEN
UMI Input
Enter UMIs in one of the following formats:
• Read name—The UMI sequence is located in the eighth colon-delimited field of the read name
(QNAME). For example, NDX550136:7:H2MTNBDXX:1:13302:3141:10799:AAGGATG+TCGGAGA
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
206
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• BAM tag—The UMI is present as an RX tag in prealigned or aligned BAM file (standard SAM
format).
• FASTQ file—The UMI is located in a third FASTQ file using the same read order as the read pairs.
To create FASTQ, append the UMI to the read name, and then specify the appropriate OverrideCycles
setting in the DRAGEN BCL conversion tool (see DRAGEN BCL Data Conversion on page 238). DRAGEN
supports UMIs with two parts each with a maximum of 8 bp and separated by +, or a single UMI with a
maximum of 15 bp.
UMI Input Correction Table
For dual, nonrandom UMIs, you can provide a predefined UMI correction table or a list of valid UMI
sequences as input. To create the UMI correction table, use a tab-delimited file, include a header, and
add the following fields.
Field Value
UMI The UMI sequence. For example, ACGTAC.
IsValid Specify if the UMI sequence is valid. Enter either: TRUE or FALSE.
NearestCodes Colon-separated list of nearest UMI sequences. For example,
ACGTAA:ACGTAT.
SecondNearestCodes Colon-separated list of second nearest sequences. For example,
ACGGAA:ACGGAT.
If customized correction table is not specified, DRAGEN uses the default table for TruSight Oncology
(TSO) UMI Reagents. Alternatively, you can provide a file for whitelisted nonrandom UMI with valid UMI
sequence one per line. DRAGEN then autogenerates a UMI correction table with hamming distance of
one.
UMI Options
• --umi-library-type—Set the batch option for different UMIs correction. Three batch modes are
available that optimize collapsing configurations for different UMI types. Use one of the following
modes:
– random-duplex—Dual, random UMIs.
– random-simplex—Single-ended, random UMIs.
– nonrandom-duplex—Dual, nonrandom UMIs. To use this option, provide a target manifest file
using --umi-metrics-interval-file.
• --umi-min-supporting-reads—Specify the number of matching UMI inputs reads required to
generate a consensus read. Any family with insufficient supporting reads is discarded. For
example, the following are the recommended settings for FFPE and ctDNA.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
207
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
– [FFPE] If the variant > 1%, use --umi-min-supporting-reads=1 with the --vc-enable-umi-solid
variant caller parameter. For more information on variant caller options, see Variant Caller
Options on page 83.
– ctDNA If the variant < 1%, use --umi-min-supporting-reads=2. with the --vc-enable-umi-liquid
variant caller parameter. For more information on variant caller options, see Variant Caller
Options on page 83
• --umi-enable—To enable read collapsing, set the --umi-enable option to true. This option is not
compatible with –enable-duplicate-marking because the UMI pipeline generates a consensus read
from a set of candidate input reads, rather than choosing the best nonduplicate read. If using the --
umi-library-type option, --umi-enable is not required.
• --umi-emit-multiplicity—Set the consensus sequence type to output. DRAGEN UMI allows you to
collapse duplex sequences from the two strands of the original molecules. Duplex sequence is
typically ~20–60% of total library, depending on library kit, input material, and sequencing depth.
Enter one of the following consensus sequence types:
– both—Output both simplex and duplex sequences. This option is the default.
– simplex—Output only simplex sequences.
– duplex—Output only duplex sequences.
• --umi-source—Specify the input type for the UMI sequence. The following are valid values: qname,
bamtag, fastq. If using --umi-source=fastq, provide the UMI sequence from FASTQ file using --
umi-fastq.
• --umi-correction-table—Enter the path to a customized correction table. By default, Local Run
Manager uses lookup correction with a built-in table for the Illumina TruSight Oncology and Illumina
for IDT UMI Index Anchor kits.
• --umi-nonrandom-whitelist—Enter the path for a customized, valid UMI sequence.
• --umi-metrics-interval-file—Enter the path for target region in BED format.
Nonrandom and Random UMI Correction
DRAGEN processes UMIs by grouping reads by UMI and alignment position. If there are sequencing
errors in the UMIs, DRAGEN can correct and detect small sequencing errors by using a lookup table or
by using sequence similarity and read counts. You specify the type of correction with the --umi-
correction-scheme option using the values “lookup,” “random,” or “none.”
For sparse sets of nonrandom UMIs, it is possible to create a lookup table that specifies which
sequence can be corrected and how to correct it. This correct file scheme works best on UMIsets
where sequences have a minimum hamming/edit distance between them. By default, DRAGEN uses
lookup correction with a built-in correct table for the Illumina TruSight Oncology and Illumina for IDT
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
208
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
UMI Index Anchor kits. Specify the path for your correction file using the --umi-library-type or --umi-
correction-file option. If you are using a different set of nonrandom UMIs, contact Illumina Technical
Support for information on generating the corresponding correction file.
In the random UMI correction scheme, DRAGEN must infer which UMIs at a given position are likely to
be errors relative to other UMIs observed at the same position. The error modes include small UMI
errors, such as one mismatch or UMI jumping or hopping artifact from library prep. DRAGEN
accomplishes this as follows.
• Groups reads by fragment alignment position.
• Within a small fuzzy window at each position, groups the reads first by the exact UMIsequence,
which forms a family.
• Estimate UMI jumping or hopping probability through insert size distribution and number of distinct
UMI at certain positions.
• Within a fuzzy window, calculates pair-wise likelihood ratio to assess if two families with different
UMI sequences and genomic positions are derived from same original molecule.
• Merges families with likelihood lower than threshold. The default threshold is 1.
Merge Duplex UMIs
Duplex UMI adapters simultaneously tag both strands of double-stranded DNA fragments. It is then
possible to identify reads resulting from amplification of each strand of the original fragment.
Consumable Prefix considers two collapsed read pairs to be the sequence of two strands of the same
original fragment of DNA if they have the same alignment position (within a fuzzy window),
complementary orientations, and their UMIs are swapped from Read 1 and Read 2. If there is only
single-ended UMI, DRAGEN compares the start-end position of families from two strands and
computes pair-wise likelihood to determine if they are likely originated from two distinct families or
should be merged as a duplex sequence. By default, DRAGEN outputs both simplex and duplex
consensus sequences. To change the consensus sequence output type, use --umi-emit-multiplicity.
Example UMI Command
The following are example UMI commands:
Generate Consensus BAM from FASTQ
The following is an example DRAGEN command for generating a consensus BAM file from input reads
with Illumina UMIs:
dragen \
-r <REF> \
-1 <FQ1> \
-2 <FQ2> \
--output-dir <OUTPUT> \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
209
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-file-prefix <PREFIX> \
--enable-map-align true \
--enable-sort true \
--umi-enable true \
--umi-correction-scheme=lookup \
--umi-min-supporting-reads 2
Use FASTQ UMI Input
To run with other random UMI library types, change --umi-library-type to random-simplex or
random-duplex.
dragen \
-r <REF> \
-1 <FQ1> \
-2 <FQ3> \
--umi-source=fastq \
--umi-fastq <FQ2> \
--output-dir <OUTPUT> \
--output-file-prefix <
PREFIX> \
--enable-map-align true \
--enable-sort true \
--umi-library-type nonrandom-duplex \
--umi-metrics-interval-file [valid target BED file]
Use Customized Correction Table
dragen \
-r <REF> \
-1 <FQ1> \
-2 <FQ2> \
--umi-correction-table <valid umi correction table> \
--output-dir <OUTPUT> \
--output-file-prefix <PREFIX> \
--enable-map-align true \
--enable-sort true \
--umi-library-type nonrandom-duplex \
--umi-metrics-interval-file <valid target BED file>
UMI Metrics
Consumable Prefix outputs an <output_prefix>.umi_metrics.csv file that describes the statistics for
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
210
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

UMI collapsing. This file summarizes statistics on input reads, how they were grouped into families,
how UMIs were corrected, and how families-generated consensus reads. The following metrics can be
useful when tuning the pipeline for your application:
• Discarded families—Any families having fewer than --umi-min-supporting-reads input or having a
different duplex/simplex status than specified by --umi-emit-multiplicity are discarded. These
reads are logged as Reads filtered out. The families are logged as Families discarded.
• UMI correction—Families may be combined in various ways. The number of such corrections are
reported as follows.
– Families shifted—Families with a very closefragment alignment coordinates (up to the
distance specified in the umi-fuzzy-window-size parameter). The default umi-fuzzy-window-
size parameter is 3.
– Families contextually corrected—Families with the same fragment alignment coordinates and
compatible UMIs are merged.
– Duplex families—Families with close alignment coordinates and complementary UMIs are
merged.
When you specify a valid path for --umi-metrics-interval-file, Consumable Prefix outputs a separate
set of on target UMI statistics that contains only families within the specified BED file.
If you need to analyze the extent to which the observed UMIs cover the full space of possible UMI
sequences, the histogram of unique UMIs per fragment position metric may be helpful. It is a zero-
based histogram, where the index indicates a count of unique UMIs at a particular fragment position,
and the value represents the number of positions with that count.
The following table describes available UMI metrics.
Metric Description
Number of reads Total number of reads.
Number of reads with
valid or correctable
UMIs
Number of reads for which the UMIs could be corrected based on the lookup
table.
Number of reads in
discarded families
Number of reads in discarded families. Families are discarded when there
are not enough raw reads to support the family.
Reads with all-G
UMIs filtered out
Number of reads filtered out due to all-G in UMI sequence.
Reads filtered out Number of reads filtered out in total either for properties or in a discarded
family.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
211
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Metric Description
Reads with
uncorrectable UMIs
Number of reads where the UMI could not be corrected.
Total number of
families
Number of simplex collapsed reads,
Families contextually
corrected
Number of families that have some contextual correction. UMI correction is
based on other families at the same mapping location.
Families shifted Number of families that have some shift correction (merging of UMI families
with very similar UMI and very close mapping locations.
Families discarded Number of families filtered out by failing min supporting reads criteria or
umi-emit type of simplex/duplex.
Families discarded by
min-support-reads
Number of families filtered out by failing min supporting reads criteria.
Families discarded by
duplex/simplex
Number of families filtered out by failing umi-emit type of simplex/duplex.
Duplex families Number of families that are merged as duplex (both strands). For
percentage, the denominator is the number of consensus pairs.
Consensus pairs
emitted
Number of collapsed reads in output BAM.
Mean family depth Average number of reads per family
Histogram of num
supporting
fragments
Number of families with zero raw reads, one raw read, two raw reads, three
raw reads, etc
Number of collapsible
regions
Number of regions.
Min collapsible region
size (num reads)
Number of reads in the most populated region.
Max collapsible
region size (num
reads)
Number of reads in the least populated region
Mean collapsible
region size (num
reads)
Average number of reads per region.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
212
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Metric Description
Collapsible region
size standard
deviation
Standard deviation of the number of reads per region.
On target number of
reads
Number of reads that overlapped with UMI target interval --umi-metrics-
interval-file.
On target number of
reads with valid or
correctable UMIs
Number of reads with a UMI that matched a UMI in the lookup table,
including error allowance, and overlapped with UMI target interval.
On target duplex
families
Number of families that are merged as duplex among all the families that
are overlapped with UMI target interval. For percentage, denominator is the
number of on-target consensus pairs.
On target mean
family depth
Average number of reads per family that overlapped with UMI target
interval.
Histogram of unique
UMIs per fragment
position
Number of positions with zero UMI sequences, one UMI sequence, two UMI
sequences, etc.
Total families in
probability model
estimation
Total number of families used in estimation of UMI jumping rate and
fragment size distribution used for probabilistic family merging.
Number of potential
Jumping Families
Total number of families that are potential UMI jumping candidates and the
corresponding ratio.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
213
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN RNA Pipeline
DRAGEN includes an RNA-seq (splicing-aware) aligner, as well as RNA specific analysis components
for gene expression quantification and gene fusion detection.
Most of the functionality and options described in Host Software Options and DNA Mapping also apply
to RNA applications. Additional RNA-specific aspects are described in this section.
Input Files
Gene Annotation File
In addition to the standard input files (reads from fastq or bam, reference genome, etc.), DRAGEN can
also take a gene annotations file as input. A gene annotations file aids in the alignment of reads to
known splice junctions,and is required for gene expression quantification and gene fusion calling.
To specify a gene annotation file, use the -a (--annotation-file) command line option. The input file
must conform to the GTF/GFF specification (http://uswest.ensembl.org/info/website/upload/gff.html).
The file must contain features of type exon, and the record must contain attributes of type gene_id and
transcript_id. An example of a valid GTF file is shown below.
chr1 HAVANA transcript 11869 14409 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000456328.2"; …
chr1 HAVANA exon 11869 12227 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000456328.2"; …
chr1 HAVANA exon 12613 12721 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000456328.2"; …
chr1 HAVANA exon 13221 14409 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000456328.2"; …
chr1 ENSEMBL transcript 11872 14412 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000515242.2"; …
chr1 ENSEMBL exon 11872 12227 . + . gene_
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
214
id "ENSG00000223972.4"; transcript_id "ENST00000515242.2"; …
chr1 ENSEMBL exon 12613 12721 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000515242.2"; …
chr1 ENSEMBL exon 13225 14412 . + . gene_
id "ENSG00000223972.4"; transcript_id "ENST00000515242.2"; …
…
Similarly, a GFF file can be used. Each exon feature must have as a Parent a transcript identifier that is
used to group exons. An example of a valid GFF file is shown below.
1 ensembl_havana processed_transcript 11869 14409
. + . ID=transcript:ENST00000456328;
1 havana exon 11869 12227
. + . Parent=transcript:ENST00000456328; …
1 havana exon 12613 12721
. + . Parent=transcript:ENST00000456328; …
1 havana exon 13221 14409
. + . Parent=transcript:ENST00000456328; …
…
The DRAGEN host software parses the file for exons within the transcripts and produces splice
junctions. The following output displays the number of splice junctions detected.
==============================================================
====
Generating annotated splice junctions
==============================================================
====
Input annotations file: ./gencode.v19.annotation.gtf
Splice junctions database file:
output/rna.sjdb.annotations.out.tab
Number of genes: 27459
Number of transcripts: 196520
Number of exons: 1196293
Number of splice junctions: 343856
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
215
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The splice junctions that are detected from the annotation file are also written to
*.sjdb.annotations.out.tab. Splice junctions below a minimum length are excluded, which helps
filter annotation artifacts that do not meet the minimum required length. This helps to lower the false
detection rate for falsely annotated junctions. This minimum annotation splice junction length is
controlled by the --rna-ann-sj-min-len option, which has a default value of 6.
Two-Pass Splice Junction Alignment
Instead of using a GTF file for annotated splice junctions, the DRAGEN software is also capable of
reading in an SJ.out.tab file (see SJ.out.tab on page 217). This file enables DRAGEN to run in a two-
pass mode, where the splice junctions discovered in the first pass (output as SJ.out.tab file) are used to
guide the mapping and alignment reads during a second run through DRAGEN. This mode of operation
is useful to increase sensitivity for spliced alignments in cases when a gene annotations file is not
readily available for the target genome.
RNA Alignment
The DRAGEN RNA pipeline uses the DRAGEN RNA-Seq spliced aligner. Mapping of short seed
sequences from RNA-Seq reads is performed similarly to mapping DNA reads. In addition, splice
junctions (the joining of noncontiguous exons in RNA transcripts) near the mapped seeds are detected
and incorporated into the full read alignments.
Alignment Output
The output files generated when running DRAGEN in RNA mode are similar to those generated in DNA
mode. RNAmode also produces extra information related to spliced alignments. Details regarding the
splice junctions are present both in the SAM alignment record and an additional file, the SJ.out.tab file.
BAM
The output BAM file meets the SAM specification and is compatible with downstream RNA-Seq
analysis tools.
RNA-Seq BAM Tags
The following BAM tags are emitted alongside spliced alignments.
• XS:A—The XS tag denotes the strand orientation of an intron. See Compatibility with Cufflinks on
page 217.
• jM:B—The jM tag lists the intron motifs for all junctions in the alignments. It has the following
definitions:
– 0: non-canonical
– 1: GT/AG
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
216
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
– 2: CT/AC
– 3: GC/AG
– 4: CT/GC
– 5: AT/AC
– 6: GT/AT
If a gene annotations file is used during the map/align stage, and the splice junction is detected as
an annotated junction, then 20 is added to its motif value.
NH:i—A standard SAM tag indicating the number of reported alignments that contains the query in the
current record. This tag may be used for downstream tools such as featureCounts.
HI:i—A standard SAM tag denoting the query hit index, with its value indicating that this alignment is
the i-th one stored in the SAM. Its value ranges from 1 … NH. This tag may be used for downstream
tools such as featureCounts.
Compatibility with Cufflinks
Cufflinks might require spliced alignments to emit the XS:A strand tag. This tag is present in the SAM
record if the alignment contains a splice junction. The values for XS:A strand tag are as follows:
‘.’ (undefined), ‘+’ (forward strand), ‘-’ (reverse strand), or ‘*’ (ambiguous).
If the spliced alignment has an undefined strand or a conflicting strand, then the alignment can be
suppressed by setting the --no-ambig-strand option to 1.
Cufflinks also expects that the MAPQ for a uniquely mapped read is a single value. This value is
specified by the --rna-mapq-unique option. To force all uniquely mapped reads to have a MAPQ equal
to this value, set --rna-mapq-unique to a nonzero value.
SJ.out.tab
Along with the alignments emitted in the SAM/BAM file, an additional SJ.out.tab file summarizes the
high confidence splice junctions in a tab-delimited file. The columns for this file are as follows:
1. contig name
2. first base of the splice junction (1-based)
3. last base of the splice junction (1-based)strand (0: undefined, 1: +, 2: -)
4. strand (0: undefined, 1: +, 2: -)
5. intron motif: 0: noncanonical, 1: GT/AG, 2: CT/AC, 3: GC/AG, 4: CT/GC, 5: AT/AC, 6: GT/AT
6. 0: unannotated, 1: annotated, only if an input gene annotations file was used
7. number of uniquely mapping reads spanning the splice junction
8. number of multimapping reads spanning the splice junction
9. maximum spliced alignment overhang
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
217
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The maximum spliced alignment overhang (column 8) field in the SJ.out.tab file is the anchoring
alignment overhang. For example, if a read is spliced as ACGTACGT------------ACGT, then the
overhang is 4. For the same splice junction, across all reads that span this junction, the maximum
overhang is reported. The maximum overhang is a confidence indicator that the splice junction is
correct based on anchoring alignments.
There are two SJ.out.tab files generated by the DRAGEN host software, an unfiltered version and a
filtered version. The records in the unfiltered file are a consolidation of all spliced alignment records
from the output SAM/BAM. However, the filtered version has a much higher confidence for being
correct due to the use of the following filters.
A splice junction entry in the SJ.out.tab file is filtered out if any of these conditions are met:
• SJ is a noncanonical motif and is only supported by < 3 unique mappings.
• SJ of length > 50000 and is only supported by < 2 unique mappings.
• SJ of length > 100000 and is only supported by < 3 unique mappings.
• SJ of length > 200000 and is only supported by < 4 unique mappings.
• SJ is a noncanonical motif and the maximum spliced alignment overhang is < 30.
• SJ is a canonical motif and the maximum spliced alignment overhang is < 12.
The filtered SJ.out.tab is recommended for use with any downstream analysis or post processing tools.
Alternatively, you can use the unfiltered SJ.out.tab and apply your own filters (for example, with basic
awk commands).
Note that the filter does not apply to the alignments present in the BAM or SAM file.
Mapping Metrics
The RNA Pipeline reports summary and per read group statistics pertaining to read mapping in the
mapping_metrics.csv file. The metrics calculation accounts for spliced alignments in RNA. The
following are some example metrics, Insert length: median, Supplementary (chimeric) alignments, etc.
Chimeric.out.junction File
If there are chimeric alignments present in the sample, then a supplementary Chimeric.out.junction file
is also output. This file contains information about split-reads that can be used to perform downstream
gene fusion detection. Each line contains one chimerically aligned read. The columns of the file are as
follows:
1. Chromosome of the donor.
2. First base of the intron of the donor (1-based).
3. Strand of the donor.
4. Chromosome of the acceptor.
5. First base of the intron of the acceptor (1-based).
6. Strand of the acceptor.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
218
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
7. N/A—not used, but is present to be compatible with other tools. It will always be 1.
8. N/A—not used, but is present to be compatible with other tools. It will always be *.
9. N/A—not used, but is present to be compatible with other tools. It will always be *.
10. Read name.
11. First base of the first segment, on the + strand.
12. CIGAR of the first segment.
13. First base of the second segment.
14. CIGAR of the second segment.
CIGARs in this file follow the standard CIGAR operations as found in the SAM specification, with the
addition of a gap length L that is encoded with the operation p. For paired end reads, the sequence of
the second mate is always reverse complemented before determining strandedness.
The following is an example entry that shows two chimerically aligned read pairs, in which one of the
mates is split, mapping segments of chr19 to chr12. Also shown are the corresponding SAM records
associated with these entries.
chr19 580462 + chr12 120876182 + 1 * * R_15448 571532
49M8799N26M8p49M26S 120876183 49H26M
chr19 580462 + chr12 120876182 + 1 * * R_15459 571552
29M8799N46M8p29M46S 120876183 29H46M
R_15448:1 99 chr19 571531 60 49M8799N26M =
580413
R_15448:2 147 chr19 580413 60 49M26S =
571531
R_15448:2 2193 chr12 120876182 15 49H26M chr19
571531
R_15459:1 99 chr19 571551 60 29M8799N46M =
580433
R_15459:2 147 chr19 580433 4 29M46S =
571551
R_15459:2 2193 chr12 120876182 15 29H46M chr19
571551
RNA Alignment Options
The aligner stage of the RNA spliced aligner uses Smith-Waterman Alignment Scoring options and
Splicing Scoring Options.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
219
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Smith-Waterman Alignment Scoring Options
Refer to Smith-Waterman Alignment Scoring Settings on page 1 for more details about the alignment
algorithm used within DRAGEN. The following scoring options are specific to the processing of
canonical and noncanonical motifs within introns.
• --Aligner.intron-motif12-pen
The --Aligner.intron-motif12-pen option controls the penalty for canonical motifs 1/2 (GT/AG,
CT/AC). The default value calculated by the host software is 1*(match-score + mismatch-pen).
• --Aligner.intron-motif34-pen
The --Aligner.intron-motif34-pen option controls the penalty for canonical motifs 3/4 (GC/AG,
CT/GC). The default value calculated by the host software is 3*(match-score + mismatch-pen).
• --Aligner.intron-motif56-pen
The --Aligner.intron-motif56-pen option controls the penalty for canonical motifs 5/6 (AT/AC,
GT/AT). The default value calculated by the host software is 4*(match-score + mismatch-pen).
• --Aligner.intron-motif0-pen
The --Aligner.intron-motif0-pen option controls the penalty for noncanonical motifs. The default
value calculated by the host software is 6*(match-score + mismatch-pen).
Splicing Scoring Options
• --Mapper.min-intron-bases
For RNA-Seq mapping, a reference alignment gap can be interpreted as a deletion or an intron. In
the absence of an annotated splice junction, the min-intron-bases option is a threshold gap length
separating this distinction. Reference gaps at least this long are interpreted and scored as introns,
and shorter reference gaps are interpreted and scored as deletions. However, alignments can be
returned with annotated splice junctions shorter than this threshold.
• --Mapper.max-intron-bases
The max-intron-bases option controls the largest possible intron that is reported, which useful for
preventing false splice junctions that would otherwise be reported. Set this option to a value that is
suitable to the species you are mapping against.
• --Mapper.ann-sj-max-indel
For RNA-seq, seed mapping can discover a reference gap in the position of an annotated intron,
but with slightly different length. If the length difference does not exceed this option, the mapper
investigates the possibility that the intron is present exactly as annotated, but an indel on one side
or the other near the splice junction explains the length difference. Indels longer than this option
and very near annotated splice junctions are not likely to be detected. Higher values may increase
mapping time and false detections.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
220
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Duplicate Marking
DRAGEN RNA can detect duplicate reads, which are defined as fragments that have both ends
mapping to the same (clipping-adjusted) position during alignment. In RNA-Seq data, the reads can
represent PCR duplicates during library prep or as a result from deep coverage of highly expressed
regions. If --enable-duplicate-marking is set to true, duplicate fragments are marked in the BAM file
and the total number of duplicate reads is reported as a mapping metric. Marking of duplicates does
not affect gene expression quantification and gene fusion calling.
MAPQ Scoring
By default, the MAPQ calculation for RNA-Seq is identical to DNA-Seq. The primary contributor to
MAPQ calculation is the difference between the best and second-best alignment scores. Therefore,
adjusting the alignment scoring parameters impacts the MAPQ estimate. These adjustments are
outlined in Smith-Waterman Alignment Scoring Settings on page 1.
The --mapq-strict-sjs option is specific to RNA, and applies where at least one exon segment is aligned
confidently, but there is ambiguity regarding possible splice junctions. When this option is set to 0, a
higher MAPQ value is returned, expressing confidence that the alignment is at least partially correct.
When this option is set to 1, a lower MAPQ value is returned, reflecting the splice junction ambiguity.
Some downstream tools, such as Cufflinks, expect the MAPQ value to be a unique value for all uniquely
mapped reads. This value is specified with the --rna-mapq-unique option. Setting this option to a
nonzero value overrides all MAPQ estimates based on alignment score. Instead, all uniquely mapped
reads have a MAPQ set to the value of --rna-mapq-unique. All multimapped reads have a MAPQvalue
of int(-10*log10(1-1/NH), where the NH value is the number of hits (primary and secondary alignments)
for that read.
Gene Fusion Detection
The DRAGEN Gene Fusion module uses the DRAGEN RNA spliced aligner for detection of gene fusion
events. It performs a split-read analysis on the supplementary (chimeric) alignments to detect potential
breakpoints. The putative fusion events then go through various filtering stages to mitigate potential
false positives. In addition to the final results, all potential candidates (unfiltered) are output, which can
be used to maximize sensitivity.
Running DRAGEN Gene Fusion
The following is an example command line for running an end to end RNA-Seq experiment.
/opt/edico/bin/dragen \
-r <HASHTABLE> \
-1 <FASTQ1> \
-2 <FASTQ2> \
-a <GTF_FILE> \
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
221
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
--output-dir <OUT_DIRECTORY> \
--output-file-prefix <PREFIX> \
--RGID <READ_GROUP_ID> \
--RGSM <Sample_NAME> \
--enable-rna true \
--enable-rna-gene-fusion true
At the end of a run, a summary of detected gene fusion events is output, which is similar to the
following example.
==================================================================
Loading gene annotations file
==================================================================
Input annotations file: ref_annot.gtf
Number of genes: 27459
Number of transcripts: 196520
Number of exons: 1196293
==================================================================
Launching DRAGEN Gene Fusion Detection
==================================================================
annotation-file:ref_annot.gtf
rna-gf-blast-pairs: blast_pairs.outfmt6
rna-gf-exon-snap: 50
rna-gf-min-anchor: 25
rna-gf-min-neighbor-dist: 15
rna-gf-max-partners: 3
rna-gf-min-score-ratio: 0.15
rna-gf-min-support: 2
rna-gf-min-support-be: 10
rna-gf-restrict-genes true
==================================================================
Completed DRAGEN Gene Fusion Detection
==================================================================
Chimeric alignments: 107923
Total fusion candidates: 38 (2116 before filters)
Time loading annotations: 00:00:08.543
Time running gene fusion: 00:00:18.470
Total runtime: 00:00:27.760
***********************************************************
DRAGEN finished normally
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
222
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Gene Expression Quantification
The DRAGEN RNA pipeline contains a gene expression quantification module, that estimates the
expression of each transcript and gene in an RNA-seq dataset. First, it internally translates the
genomic mapping of each read (read pair) to the corresponding transcript mappings. Then it uses an
Expectation-Maximization (EM) algorithm to infer the transcript expression values that best match all
the observed reads. The EM algorithm can also model GC-bias and correct for it in the reported
quantification results.
DRAGEN Single-Cell RNA Pipeline
The DRAGEN Single-Cell RNA (scRNA) Pipeline can process multiplexed single-cell RNA-Seq data sets
from reads to a cell-by-gene UMI count gene expression matrix. The pipeline is compatible with library
designs that have one read in a fragment match to a transcript and the other containing a cell-barcode
and UMI. The pipeline includes the following functions:
• RNA-Seq (splice-aware) alignment and matching to annotated genes for the transcript reads.
• Cell-barcode and UMI error correction for the barcode read.
• UMI counting per cell and gene to measure gene expression.
• Sparse matrix output and QC metrics.
The functionality and options related to alignment and gene annotation are identical to the RNA-Seq
pipeline. For information, see DRAGEN RNA Pipeline on page 214. Other RNA-Seq modules, such as
gene fusion calling or transcript-level gene expression quantification are not supported for Single-Cell
RNA.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
223
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Input Files
Alignment Reference
Use a standard DRAGEN RNA reference genome or hashtable for the DRAGENscRNA Pipeline. For
example, build using --ht-build-rna-hashtable=true). The pipeline also requires a gene
annotation file in GTF format, provided with the --annotation (-a) option.
Read Input
The DRAGEN scRNA Pipeline requires both the transcript sequence and the barcode+UMI sequence
for each fragment (read) as input. The transcript sequence is aligned to the genome to determine the
expressed gene, the barcode+UMI sequence is split into the single-cell barcode to identify the unique
cell, and a single-cell UMI for unique molecule quantification.
When starting from FASTQ, you can either include the UMI in the read name or provide separate UMI
FASTQ files.
UMI in Read Name
Provide the transcriptome reads as a single-end FASTQ file with the Barcode+UMI sequence in the
eighth field of the read-name line. Separate sequences using a colon. The following example uses
read2 (sample.R2.fastq.gz) as the transcript read.
@D00626:253:CAT5WANXX:4:1302:8433:85343:GAAACTCGTTCAGCGCTCATCTCGGC
ACAGGTCAGCTGGGAGCTTCTGCCCCCACTGCCTAGGGACCAACAGGGGCAGGAGGCAGTCACTGACCCCGAGA
AGTTTGCATCCTGCACAGCTAGAGATC
+
CCCCBFGGGGGGGGGGGGGGGGGGBDCGGGE>1BGDFGG0F@FBGGGBCDGGGGGGGGGGGGGGGGGGGGGGGD
GGGGGGGGGGGGGFGGGGFGEGGGGGE
In the example, the GAA sequence is the barcode+UMI read and the ACAG sequence is the
transcriptome read.
These FASTQ files can be generated by bclConvert and bcl2fastq using the UMI settings to define the
single-cell barcode+UMI read. If using bclConvert, enter the barcode/UMI information using the
OverrideCycles setting. For more information, see the BCL Convert Software Guide (document #
1000000094725).
bclConvert refers to the entire single-cell barcode+UMI sequence as UMI.
Enter the following command line option to use the generated FASTQ files from bclConvert:
dragen -1 <file name> --umi-source=qname
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
224
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The option is also compatible with the --fastq-list input options and with read input from BAM
files.
Separate UMI FASTQ Files
An alternative option is to provide the transcript and barcode+UMI sequences as two separate FASTQ
files. One file contains only the transcriptome reads and one contains the corresponding barcode-
reads in the same order. This file is similar to how read-pairs are normally handled. If using separate
UMI files, the sequencing system run setup and bclConvert are not aware of the UMI at all and treat it
as normal read sequence by default.
Enter the following command line option to use the separate UMI FASTQ files:
dragen -1 <file name> --umi-fastq=<file name> --umi-source=fastq
To use this method with multiple FASTQ files, do as follows.
1. Enter the barcode+UMI FASTQ files as read1 in the fastq-list file, and then enter the
transcriptome read FASTQ files matching the default fastq_list.csv generated by bclConvert
as read2.
2. Enter the following command:
dragen --fastq-list fastq_list.csv --umi-source=read1
Using Multiple Libraries
The scRNA pipeline can process a single biological sample per DRAGENrun. To process multiple single-
cell libraries together, split the single sample into multiple single-cell libraries with a unique set of cells
in each DRAGEN keeps the cells (barcodes and UMIs) from each library separate and provides merged
outputs across all. Read groups are used to specify the library for each FASTQ file using the RGLB
attribute.
Single-Cell RNA Settings
To use the scRNA workflow, enter --enable-rna=true --enable-single-cell-rna=true. This
section includes information on additional scRNA settings.
Barcode Options
By default the scRNA workflow assumes that the overall barcode/UMI sequence is made up of a single-
cell barcode (possibly split into multiple blocks) and a single UMI. Enter the following command to
identify the location of the single-cell barcode and single-cell UMI in the barcode read:
--single-cell-barcode-position <blockPos>[+<blockPos2>+<blockPos3>...] --
single-cell-umi-position <blockPos>
blockPos describes the offset of the first and last inclusive base of the block and is formatted as
<startPos>_<endPos>. For example for a library with a 16 bp cell-barcode followed by a 10 bp UMI,
enter: --single-cell-barcode-position 0_15 --single-cell-umi-position 16_25. For a
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
225
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
library with the cell-barcode split into three blocks of 9 bp separated by fixed linker sequences and an 8
bp UMI, enter --single-cell-barcode-position=0_8+21_29+43_51 --single-cell-umi-
position=52_59.
Cell filtering
DRAGEN uses a threshold on the number of unique UMIs per cell barcode to determine which barcodes
likely correspond to single-cells in the original sample from background noise. The threshold is
determined based on the distribution of UMIs per barcode and an expected number of true cells in the
sample. For more information on how cell filtering is performed, see Cell Filtering on page 227 .
• single-cell-number-cells—[Optional] Set the expected number of cells. The default is 3000. Adjust
only if the number of cells is far from the default.
• single-cell-threshold— Set the UMI count threshold value or use the value fixed. If using fixed, the
UMI count threshold is set so that the expected number of cells pass, rather than estimated
dynamically. The exact number of passing cells might be slightly larger than the number of single-
cells because of the connection between the UMI count and the different cell barcodes.
To set a specific number of cells ahead of time, use the following command:
--single-cell-threshold=fixed --single-cell-number-cells=X
The command forces the UMI threshold value to pass the top X cells and any extra cells with the
same number of unique UMIs.
Additional Options
The following are additional options you can use to configure the Single-Cell RNA Pipeline settings.
• rna-libary-type—Set the orientation of transcript reads relative to the genomes. Enter SF for
forward, SR for reverse, or U for unstranded. The default is SF.
• single-cell-count-introns—Include intronic reads in gene expression estimation. The default false.
• qc-enable-depth-metrics—Set to false to disable depth metrics for faster run times. The default
is true.
• bypass-anchor-mapping—Set to true to disable RNA anchor (two-pass) mapping for increased
performance. The default false.
Command-line Example
The following is an example command line to run the DRAGEN Single Cell RNA Pipeline.
dragen --enable-rna=true --enable-single-cell-rna=true --umi-source=fastq
--single-cell-barcode 0_15 --single-cell-umi 16_25 -r reference_
genomes/Mus_musculus/mm10/DRAGEN/8 -a reference_genomes/Mus_
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
226
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
musculus/mm10/gtf/gencode.vM23.annotation.gtf.gz -1 lib1_S7_L001_R2_
001.fastq.gz --umi-fastq lib1_S7_L001_R1_001.fastq.gz --RGID=1 --
RGSM=sample1 --output-dir=/staging/out --output-file-prefix=sample1
Cell Filtering
DRAGEN uses a UMI count threshold to separate cell-barcodes corresponding to single-cells in the
original sample from background noise. Cell-barcodes with UMI counts beneath the threshold are
considered background noise. To set the threshold, DRAGEN sorts all cell-barcodes by their UMI count
in descending order and then determines the counts of the following two reference cell-barcodes:
• topCount—The maximum corresponding to the count of the cell-barcode at the first percentile of
the UMI count distribution.
• minCount—An expected minimum corresponding to the count of the last expected cell. The
minCount is represented as the _N_th cell, where N is the number of expected cells.
The final threshold is then set as max(topCount * 0.1, minCount * 0.5.
Barcode Error Correction
Cell-barcode sequences from the input reads are error corrected based on the frequency with which
each one is seen and an optional whitelist of expected cell-barcode sequences. A cell-barcode
sequence is considered a neighbor of another cell-barcode if there is at most one mismatch. A cell-
barcode sequence is corrected to its neighbor in the following circumstances. When corrected, all reads
with the cell-barcode are assigned instead to the neighboring cell-barcode. The sequence error
correction scheme is similar to the directional algorithm described in (Smith, Heger and Sudbery,
2020)¹.
• The neighboring cell-barcode is at least two times more frequent across all input reads.
• The neighboring cell-barcode is on the cell-barcode whitelist, but the original cell-barcode is not.
To avoid overcounting UMIs based on sequence errors, UMI error correction is performed among all
reads with the same cell-barcode mapping to the same gene. UMI sequences that are likely errors of
another UMI are not counted.
¹Smith, T., Heger, A. and Sudbery, I., 2020.UMI-Tools: Modeling Sequencing Errors In Unique
Molecular Identifiers To Improve Quantification Accuracy. [PDF] Cold Spring Harbor Laboratory Press.
Available at: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5340976/> [Accessed 15 October
2020].
Outputs
Single-cell RNA outputs are found in the standard DRAGEN output location using the prefix scRNA.
Counts
The following three files provide information per-cell gene expression level in matrix market (*.mtx)
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
227
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

format:
• <prefix>.scRNA.matrix.mtx
Count of unique UMIs for each cell/gene pair in sparse matrix format.
• <prefix>.scRNA.barcodes.tsv
Cell-barcode sequence for each cell from the matrix.
• <prefix>.scRNA.genes.tsv
Gene name and ID for each gene in the matrix. This includes all cell barcodes. The subset
corresponding to passing cells can be found under the Filter column in
scRNA.barcodeSummary.tsv.
Alignments
Alignments of the transcript reads are sorted by coordinate and output as a BAM file. Each alignment is
annotated with an XB tag containing the cell barcode and an RX tag containing the UMI. The
alignments use the original sequences without any errors corrected. Fragments that did not have an
associated barcode read, for example fragments trimmed on the input data, do not XB and RX tags.
Overall Metrics
The <prefix>.scRNA.metrics.csv file contains per sample scRNA metrics.
Barcode Read Metrics
Metric Description
Invalid barcode read Overall barcode sequence (cell barcode + UMI) failed basic
checks. For example, the barcode read was missing or too
short.
Error free cell-barcode Reads with cell-barcode sequences that were not altered
during error correction. For example, if the read was an
exact match to the whitelist.
Error corrected cell-barcode Reads with cell-barcode sequences successfully corrected
to a valid sequence.
Filtered cell-barcode Reads with cell-barcode sequences that could not be
corrected to a valid sequence. For example, the sequence
does not match whitelist with at most one mismatch.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
228
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Transcript Read Metrics
Metric Description
Unique exon match Reads match to exons of exactly one gene.
Unique intron match Reads do not match exons, but introns of exactly one gene.
For example, if using the command --single-cell-
count-introns=true.
Ambiguous match Reads match to multiple genes.
Wrong strand Reads overlap a gene on the opposite strand defined by
library type.
Mitochondrial reads Reads map to the mitochondrial example, if there is a
matching gene.
No gene Reads do not match to any gene. Includes intronic reads
unless using --single-cell-count-introns=true .
Filtered multimapper Reads excluded due to multiple alignment positions in the
genome.
UMI Count Metrics)
Metric Description
Total counted reads Reads with valid cell-barcode and UMI that match a unique
gene.
Reads with error-corrected UMI Counted reads where the UMI was error-corrected to
match another similar UMI sequence.
Reads with invalid UMI Reads that were not counted due to invalid UMI sequence.
For example, pure homopolymer reads or reads containing
Ns.
Sequencing saturation Fraction of reads with duplicate UMIs. 1 - (UMIs / Reads).
Unique cell-barcodes Overall number of unique cell-barcode sequences in
counted reads only.
Unique UMIs Overall number of unique cell-barcode and UMI
combinations counted.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
229
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Cell Metrics
Metric Description
UMI threshold for passing cells Number of UMIs required for a cell-barcode to pass
filtering.
Passing cells Number of cell-barcodes that passed.
Fraction genic reads in cells Counted reads assigned to cells that passed.
Median reads per cells Total counted reads per cell that passed the filters.
Median UMIs per cells Total counted UMIs per cell that passed the filters.
Median genes per cells Genes with at least one UMI per cell that passed the filters.
Total genes detected Genes with at least one UMI in at least one cell that passed
the filters.
Per-Cell Metrics
The <prefix>.scRNA.barcodeSummary.tsv contains summary statistics for each unique cell-
barcode per cell after error correction.
Metric Description
ID Unique numeric ID for the cell-barcode. The ID matches
the line in UMI count matrix (*.mtx) output.
Barcode The cell-barcode sequence.
TotalReads Total reads with the cell-barcode sequence. This includes
error corrected reads.
GeneReads Reads counted towards a gene.
UMIs Total number of UMIs in counted reads.
Genes Unique genes detected.
MitochondrialReads Reads mapped to mitochondrial genome.
Filter The following are the available filter values:
• PASS—Cell-barcode passes the filter.
• LOW—UMI count is below the threshold.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
230
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

DRAGEN Methylation Pipeline
The epigenetic methylation of cytosine bases in DNA can have a dramatic effect on gene expression,
and bisulfite sequencing is the gold standard for detecting epigenetic methylation patterns at single-
base resolution. This technique involves chemically treating DNA with sodium bisulfite, which converts
unmethylated cytosine bases to uracil, but does not alter methylated cytosines. Subsequent PCR
amplification converts any uracils to thymines.
A bisulfite sequencing library can either be nondirectional or directional. For nondirectional, each
double-stranded DNA fragment yields four distinct strands for sequencing, post-amplification, as
shown in the following figure:
Figure 15 Nondirectional Bisulfite Sequencing
– Bisulfite Watson (BSW), reverse complement of BSW (BSWR),
– Bisulfite Crick (BSC), reverse complement of BSC (BSCR)
For directional libraries, the four strand types are generated, but adapters are attached to the DNA
fragments such that only the BSW and BSC strands are sequenced (Lister protocol). Less commonly,
the BSWR and BSCR strands are selected for sequencing (eg, PBAT).
BSW and BSC strands:
• A, G, T: unchanged
• Methylated C remains C
• Unmethylated C converted to T
BSWR and BSCR strands:
• Bases complementary to original Watson/Crick A, G, T bases remain unchanged
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
231
• G complementary to original Watson/Crick methylated C remains G
• G complementary to original Watson/Crick unmethylated C becomes A
Standard DNA sequencing is used to produce sequencing reads. Reads containing more methylated
C’s or G’s complementary to methylated C’s are less drastically affected by the bisulfite treatment, and
have a higher likelihood of mapping to the reference than reads with more bases affected. The
standard protocol to minimize this mapping bias is to perform multiple alignments per read, where
specific combinations of read and reference genome bases are converted in-silico prior to each
alignment run. Each alignment run has a set of constraints and base-conversions that corresponds to
one of the bisulfite+PCR strand types expected from the protocol. By comparing the per read (pair)
alignments across runs, DRAGEN determines the best alignment, and most likely, strand type for each
read or read pair. DRAGEN can then use the alignments and strands for downstream methylation
calling.
DRAGEN Methylation Calling
Different methylation protocols require the generation of two or four alignments per input read,
followed by an analysis to choose a best alignment and determine which cytosines are methylated.
DRAGEN can automate this process, generating a single output BAM file with Bismark-compatible tags
(XR, XG, and XM) that can be used for methylation calling and other downstream workflow.
When the --methylation-protocol option is set to a valid value other than none, DRAGEN automatically
produces the required set of alignment runs, each with the appropriate base conversions on the reads,
base conversions on the reference, and constraints on whether reads must be forward-aligned or
reverse complement (RC) aligned with the reference.
The following options are automatically configured.
• --preserve-map-align-order true \
• --generate-md-tags true \
• --Aligner.global 1 \
• --Aligner.no-unpaired 1 \
• --Aligner.aln-min-score 0 \
• --Aligner.min-score-coeff -0.2 \
• --Aligner.match-score 0 \
• --Aligner.mismatch-pen 4 \
• --Aligner.gap-open-pen 6 \
• --Aligner.gap-ext-pen 1 \
• --Aligner.supp-aligns 0 \
• --Aligner.sec-aligns 0
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
232
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Because global alignments (end-to-end in the reads) are generated, DRAGEN recommends trimming
any artifacts introduced by library prep and adapter sequences.
When --enable-methylation-calling is set to true, DRAGEN analyzes the multiple alignments to produce
a single methylation-tagged BAM file. When --enable-methylation-calling is set to false, DRAGEN
outputs a separate BAM file per alignment run.
The following table describes these alignment runs:
Protocol BAM Reference Read 1 Read 2
Orientation
Constraint
directional
1 C->T C->T G->A Forward-only
2 G->A C->T G->A RC-only
nondirectional, or directional-complement
1 C->T C->T G->A Forward-only
2 G->A C->T G->A RC-only
3 C->T G->A C->T RC-only
4 G->A G->A C->T Forward-only
PBAT
3 C->T G->A C->T RC-only
4 G->A G->A C->T Forward-only
In directional protocols, the library is prepared such that only the BSW and BSC strands are sequenced.
Thus, alignment runs are performed with the two combinations of base conversions and orientation
constraints best suited for these strands (directional runs 1 and 2 above).
With nondirectional protocols, reads from each of the four strands are equally likely, so alignment runs
must be performed with two more combinations of base conversions and orientation constraints
(nondirectional runs 3 and 4 above).
In PBAT protocols, the library is prepared so only the BSWR and BSCR strands are sequenced. Only two
alignment runs are performed with the combinations of base conversions and orientation constraints
best suited for these strands (runs 3 and 4).
The directional-complement protocol can also be used for PBAT or similar libraries where mainly the
BSWR and BSCR strands are sequenced. With this protocol, all four aligner runs are performed, but
relatively few good alignments are expected from the runs for the BSW and BSC strands, so DRAGEN is
automatically tuned to a faster analysis mode for those runs.
For any protocol, you must use a reference hash table that was produced with --ht-methylated true.
For more information, see Pipeline Specific Hash Tables on page 21.
The following is an example DRAGEN command line for the directional protocol:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
233
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

dragen --enable-methylation-calling true \
--methylation-protocol directional \
--ref-dir /staging/ref/mm10/methylation --RGID RG1 --RGCN CN1 \
--RGLB LIB1 --RGPL illumina --RGPU 1 --RGSM Samp1 \
--intermediate-results-dir /staging/tmp \
-1 /staging/reads/samp1_1.fastq.gz \
-2 /staging/reads/samp1_2.fastq.gz \
--output-directory /staging/outdir \
--output-file-prefix samp1_directional_prot
Methylation-Related BAM Tags
When --enable-methylation-calling is set to true, DRAGEN analyzes the alignments produced for the
configured --methylation-protocol, and generates a single output BAM file that includes methylation-
related tags for all mapped reads.As in Bismark, reads without a unique best alignment are excluded
from the output BAM.The added tags are as follows:
Tag Brief Description Description
XR:Z Read conversion For the best alignment, which
base-conversion was performed
on the read: CT or GA.
XG:Z Reference conversion For the best alignment, which
base-conversion was performed
on the reference: CT or GA
XM:Z Methylation call A byte-per-base methylation
string.
The XM:Z (methylation call) tag contains a byte corresponding to each base in the read’s
sequence.There is a period (‘.’) for each position not involving a cytosine, and a letter at cytosine
positions.The letter indicates the context (CpG, CHG, CHH, or unknown). The case indicates
methylation, with upper-case positions being methylated and lower-case unmethylated.The letters
used at cytosine positions are as follows:
Character Methylated? Context
. not cytosine not cytosine
z No CpG
Z Yes CpG
X No CHG
X Yes CHG
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
234
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Character Methylated? Context
h No CHH
H Yes CHH
u No Unknown
U Yes Unknown
Methylation Cytosine and M-Bias Reports
To generate a genome-wide cytosine methylation report, set --methylation-generate-cytosine-report
to true.The position andstrand of each C in the genome are givenin the first three fields of the report.
Arecord with a'-' in the strand field is foraG in the reference FASTA. The counts of methylated and
unmethylated C's covering the position are given in the fourth and fifth fields, respectively. The C-
context in the reference (CG, CHG, or CHH) is given in the sixth field, and the trinucleotide sequence
context is given in the last field (eg, CCC, CGT, CGA, etc). The cytosine report only includes records
forThe following is an example cytosine report record:
chr2 24442367 + 18 0 CG CGC
To generate an M-bias report, set --methylation-generate-mbias-report to true. This report contains
three tables for single-ended data with one table for each C-context, and six tables for paired-end
data. Each table is a series of records, with one record per read base position. For example, the first
record for the CHG table contains the counts of methylated C's (field 2)and unmethylatedC's (field 3)
thatoccur in the first read base position, restricting to thosereads in which the firstbase is aligned to a
CHG location in the genome. Each record of a table alsoincludes the percent methylated C bases (field
4)and the sum ofmethylated and unmethylated C counts (field 5).
The following is an example M-bias record for read base position 10:
10 7335 2356 75.69 9691
For data sets with paired-end reads that overlap, both the Cytosine and M-bias reports skip reporting
any C’s in the second read that overlaps the first read. In addition, 1-based coordinates are used for
positions in both reports.
To match thebismark_methylation_extractor cytosine and M-bias reports generated by Bismark
version 0.19.0, set the --methylation-match-bismark option to true. The ordering of records in Bismark
and DRAGENcytosine reports may differ. DRAGEN reports are sorted by genomic position.
Using Bismark for Methylation Calling
The recommended approach to methylation calling is to automate the multiple required alignment runs,
and add the XM, XR, XG tags. To generate a separate BAM file for each of the constraints and
conversions needed by the methylation protocol, set --enable-methylation-calling to false. The BAM
files can be used as input into Bismark for methylation calling. Contact Illumina Technical Support for
more information on this use case if desired.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
235
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
In this mode, a single DRAGEN run produces multiple BAM files in the directory specified by --output-
directory, each containing alignments in the same order as the input reads. It is not possible to enable
sorting or duplicate marking for these runs. Alignments include MD tags and have /1 or /2 appended to
the names of paired-end reads for Bismark-compatibility. The BAM files use the following naming
conventions:
• Single-end reads—output-directory/output-file-prefix.{CT,GA}read{CT,GA}reference.bam
• Paired-end-reads—output-directory/output-file-prefix.{CT,GA}read1{CT,GA}read2
{CT,GA}reference.bam,
Where output-directory and output-file-prefix are specified by the corresponding options, and CT and
GA correspond to the base conversions listed in the table above.
Bismark does not have a directional-complement mode, but you can process such samples using
Bismark’s nondirectional mode with the expectation that runs 1 and 2 produce very few good
alignments.
Using TAPS Support
TET-Assisted Pyridine Borane Sequencing (TAPS) is a new assay which directly converts methylated C
to T, whereas typical bisulfite conversion converts unmethylated C to T. This approach preserves
genomic complexity and uses less destructive chemicals to enable lower input DNA.
To enable analysis of FASTQ data generated through TAPS, set --methylation-TAPS to true. By
default, the option is false. This option is performed only during the alignment step and is not necessary
when generating methylation cytosine and M-Bias reports from an existing BAM.
Sort and Duplicate Reads Options
DRAGEN supports sorting and duplicate marking/removal for methylated reads during the alignment
phase. To enable sort and duplicate read options, perform two separate runs as follows.
1. Set the options for the first run as follows.
• To generate sorted alignment output (in BAM format), set --enable-sort to true.
• To detect duplicate reads, set --enable-duplicate-marking to true.
• [Optional]To remove duplicate reads, set --remove-duplicates to true.
• Set --methylation-generate-cytosine-report and --methylation-generate-mbias-
report to false.
These options behave the same as in DNA alignment. For example, if --enable-duplicate-
marking is set to true, --enable-sort is true.
2. Set the options for the second run as follows.
• Use the sort/markdup/dedup alignment output from the previous run for -b/--bam-input.
• Set --methylation-reports-only to true.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
236
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• Set --enable-sort to false.
• To generate cytosine report, set --methylation-generate-cytosine-report to true.
• To generate mbias report, set --methylation-generate-mbias-report to true.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
237
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Tools and Utilities
DRAGEN BCL Data Conversion
BCL format is the native output format of Illumina sequencing systems and consists of a directory
hierarchy containing data files and metadata. The data files are organized according to the flow cell
layout of the sequencing system. The software converts this data to sample separated FASTQ files.
DRAGEN provides BCL conversion software that uses hardware acceleration on the DRAGEN platform,
which results in improved run times compared to a pure software execution. To run the conversion
software, use the --bcl-input-directory <BCL_ROOT>, --output-directory <DIR>, and --bcl-conversion-
only true options.
The DRAGEN BCL conversion is designed to output FASTQ files that match bcl2fastq2 v2.20 output.
DRAGEN BCL conversion supports the following features.
• Demultiplexing samples by barcode with optional mismatch tolerance.
• Adapter sequence masking or trimming with adjustable matching stringency.
• UMI sequence tagging and optional trimming.
• Output of FASTQ files for index reads.
• [Optional] Combine all lanes to the same FASTQ output files.
• High sample count support (100,000).
• UMI sequences in index reads.
• Eliminate skew as the result of adapter sequence trimming by using the MinimumAdapterOverlap
setting.
• Outputs metrics to detect index-hopping.
Command Line Options
The following example command contains the required BCL conversion options:
dragen --bcl-conversion-only true --bcl-input-directory <...> --output-
directory <...>
The following additional options can be specified on the command line:
• --sample-sheet—Specifies the path to SampleSheet.csv file. --sample-sheet is optional if the
SampleSheet.csv file is in the --bcl-input-directory directory.
• --run-info—Sets the path to the RunInfo.xml file. By default, the file is located in the --bcl-input-
directory directory.
• --strict-mode—If set to true, DRAGEN aborts if any files are missing. The default is false.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
238
• --first-tile-only—If set to true, DRAGEN only converts the first tile of input (for testing and
debugging). The default is false.
• --bcl-only-lane <#>—Convert only the specified lane in this conversion run.
• -f—Convert to output directory even if the directory exists (force).
• --bcl-use-hw false—Do not use DRAGEN FPGA acceleration during BCL conversion. This allows
concurrent execution of BCL conversion with DRAGEN analysis.
• --bcl-sampleproject-subdirectories true—Output FASTQ files to subdirectories based on sample
sheet Sample_Project column.
• --no-lane-splitting true—Output all lanes of a flow cell to the same FASTQ files consecutively.
• --bcl-only-matched-reads true—Disable outputting unmapped reads to files marked as
Undetermined.
The following additional options can be used to manually control performance. Use of these options
might reduce performance or result in analysis failure. Contact Illumina Technical Support if issues
occur.
• --shared-thread-odirect-output true —Switch to an alternate file output method that is optimized
for sample counts greater than 100,000. This option is not recommended for lower sample counts
and/or if using distributed file system targets such as GPFS or Lustre.
• --bcl-num-parallel-tiles <#>—Number of tiles processed in parallel. The default is determined
dynamically.
• --bcl-num-conversion-threads <#>—Number of conversion threads per tile. The default is
determined dynamically.
• --bcl-num-compression-threads <#>—Number of CPU threads for compressing FASTQ output.
The default is determined dynamically.
• --bcl-num-decompression-threads <#>—Number of CPU threads for decompressing input BCL
files. The default is determined dynamically.
The input BCL root directory and the output directory must be specified. The specified input path is
three levels higher than the BaseCalls directory and should contain the RunInfo.xml file.
To specify the directory to store output FASTQ files, use the --output-directory option.
Sample Sheet Options
In addition to the command line options that control the behavior of BCL conversion, you can use the
[Settings] section in the sample sheet configuration file to specify how the samples are processed. The
following are the sample sheet settings for BCL conversion.
DRAGEN does not support the following sample sheet settings from bcl2fastq
• FindAdapterWithIndels
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
239
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

• ReverseComplement
Option Default Value
Descripti
on
AdapterBehavior trim trim, mask Whether
adapter
should be
trimmed
or
masked.
AdapterRead1 None Read 1 adapter sequence
containing A, C, G, or T
The
sequence
to trim or
mask
from the
end of
Read 1.
AdapterRead2 None Read 2 adapter
sequence containing A,
C, G, or T
The
sequence
to trim or
mask
from the
end of
Read 2.
AdapterStringency 0.9 Float between 0.5 and
1.0
The
stringenc
y for
matching
the read
to the
adapter
using the
sliding
window
algorith
m.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
240
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Default Value
Descripti
on
BarcodeMismatchesInde
x1
1 0, 1, or 2 The
number
of
allowed
mismatch
es
between
the first
Index
Read and
index
sequenc
e.
BarcodeMismatchesInde
x2
1 0, 1, or 2 The
number
of
allowed
mismatch
es
between
the
second
Index
Read and
index
sequenc
e.
MinimumTrimmedReadL
ength
The minimum of 35 and
the shortest non-indexed
read length.
0 to the shortest non-
indexed read length
Reads
trimmed
below
this point
become
masked
at that
point.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
241
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Default Value
Descripti
on
MinimumAdapterOverlap 1 1, 2, or 3 Do not
trim
detected
adapter
sequence
s shorter
than this
value.
MaskShortReads The minimum of 22 and
MinimumTrimmedReadLe
ngth.
0 to
MinimumTrimmedReadL
ength
Reads
trimmed
below
this point
become
masked
out.
TrimUMI 1 0 or 1 If set to 0,
UMI
sequence
s are not
trimmed
from
output
FASTQ
reads.
The UMI
is still
placed in
sequence
header.
CreateFastqForIndexRea
ds
0 0 or 1 If set to 1,
output
FASTQ
files for
index
reads and
genomic
reads.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
242
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Default Value
Descripti
on
OverrideCycles None Y: Specifies a sequencing
read
I: Specifies an indexing
read
U: Specifies a UMI length
to be trimmed from read
String
used to
specify
UMI
cycles
and mask
out cycles
of a read.
Override Cycles
The OverrideCycles mask elements are semicolon separated. For example:
OverrideCycles,U7N1Y143;I8;I8;U7N1Y143
DRAGEN supports flexible UMI processing during BCL conversion to support more third-party assays,
including UMI sequences in index reads and multiple UMI regions per read. UMI sequences are
trimmed from FASTQread sequences and placed in the sequence identifier for each read, as normal.
The following are examples of OverrideCycles settings using 2x151 reads:
Setting Description
OverrideCycles,U7N1Y143;I8;I8;U7N1Y143 UMI is comprised of the first 7 bps of each
genomic read, linked by 1 bps of ignored
sequence. This is the format for Illumina
nonrandom UMIs, used in the following products:
• TruSight Oncology 170 RUO
• TruSight Oncology 500 RUO
• IDT for Illumina - UMI Index Anchors
OverrideCycles,Y151;I8;U10;Y151 Index Read 2 is a 10 bps UMI. This is the format
for Agilent XT HS.
OverrideCycles,Y151;I8U9;I8;Y151 Index Read 1 contains both an index and a 9 bps
UMI. This is the format for IDT Dual Index
Adapters with UMIs.
OverrideCycles,U3N2Y146;I8;I8;U3N2Y146 UMI is comprised of the first 3 bps of each
genomic read, linked by 2 bps of ignored
sequence. This is the format for UMIs in
SureSelect XT HS 2 and IDT xGen Duplex Seq
Adapter.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
243
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Setting Description
OverrideCycles,Y151;I8;I8;U10N12Y127 UMI is at the beginning of Read 2, attached with
a linker sequence of length 12.
No lane splitting
When using --no-lane-splitting true, DRAGEN FASTQ file name convention and FASTQ contents match
bcl2fastq2 for the same feature.
DRAGEN only supports this mode when the Lane column is not specified in the sample sheet to make
sure that all samples are present in all lanes in the same order listed. This is generally expected for flow
cells with no fluidic boundaries between lanes.
BCL Metrics
DRAGEN BCL conversion outputs metrics in CSV format to the Reports/ output subfolder. Information
provided includes metrics files for demultiplexing, adapter sequence trimming, index-hopping (for
unique-dual indexes only), and the top unknown barcodes for each lane. In addition, the sample sheet
and RunInfo.xml file used during conversion is copied into the Reports/ subdirectory for reference.
Demultiplex Output File
The following information is included in the Demultiplex_Stats.csv output file.
Column Description
Lane The lane for each metric.
SampleID The contents of Sample_ID in the sample sheet for this sample.
Index The contents of index in sample sheet for this sample. For dual-index, the
value concatenated with index2.
# Reads The total number of pass-filter reads mapping to this sample for the lane.
# Perfect Index
Reads
The number of mapped reads with barcodes that match the indexes provided
in the sample sheet.
# One Mismatch
Index Reads
The number of mapped reads with barcodes matched with one base
mismatched.
# of >= Q30 Bases
(PF)
The total number of bases mapped to this sample with a quality score greater
than or equal to 30.
Mean Quality Score
(PF)
The mean quality score of all bases mapping to this sample.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
244
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Adapter Metrics Output File
The following information is included in the Adapter_Metrics.csv output file.
Column Description
Lane The lane for each metric.
Sample_ID The contents of Sample_ID in the sample sheet for this sample.
index The contents of index in sample sheet for this sample.
index2 The contents of index2 in the sample sheet for this sample.
R1_
AdapterBases
The total number of bases trimmed as adapter from read 1 reads.
R1_
SampleBases
The total number of bases not trimmed from read 1.
R2_
AdapterBases
The total number of bases trimmed as adapter from read 2 reads.
R2_
SampleBases
The total number of bases not trimmed from read 2.
# Reads The total number of pass-filter reads mapping to this sample in this
lane.
Index Hopping Counts Output File
For unique dual index inputs, the Index_Hopping_Counts.csv file provides the number of reads
mapping to every possible combination of provided index and index2 values, including via mismatch
tolerance. The metrics provide visibility into any index-hopping behavior that might be occurring. The
samples with both index and index2 values present in the sample sheet are present in the index
hopping file for reference. The following information is included in the Index_Hopping_Counts.csv
output file.
Column Description
Lane The lane for each metric.
SampleID If the index combination corresponds to a sample, the
contents of Sample_IDin the sample sheet for this
sample.
index The contents of index in sample sheet for the sample.
index2 The contents of index 2i n sample sheet for the sample.
# Reads The total number of pass-filter reads mapping to the
index and index2 combination.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
245
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Top Unknown Barcodes Output File
The Top_Unknown_Barcodes.csv file lists the most commonly-encountered barcode sequences in
the flow cell input that are not listed in the sample sheet. The 100 most common unlisted sequences
are listed, along with any other sequences with a frequency equivalent to the 100th most commonly
encountered sequence. The following information is included in the Top_Unknown_Barcodes.csv
output file.
Column Description
Lane The lane for each metric.
index The first index value of this unlisted sequence
index2 The second index value of this unlisted sequence
# Reads The total number of pass-filter reads mapping to the index
and index2 combination
FASTQ List Output File
The fastq_list.csv output file is located in the output folder with the FASTQ files. The files
provides the associations between the sample indexes, lane, and the output FASTQ file names. For
information on running DRAGEN using fastq_list.csv, see, lane, and the output FASTQ file names.
The columns of each row are documented below, along with example entries from a test run. For more
information on running DRAGEN using fastq_list.csv, see FASTQ CSV File Format.
Column Description
RGID Read Group
RGSM Sample ID
RGLB Library
Lane Flow cell lane
Read1File Full path to a valid FASTQ input file
Read2File Full path to a valid FASTQ input file. Required for paired-
end input. If not using paired-end input, leave empty,
The following is an example fastq_list.csv output file.
RGID,RGSM,RGLB,Lane,Read1File,Read2File
AACAACCA.ACTGCATA.1,1,UnknownLibrary,1,/home/user/dragen_bcl_out/1_S1_
L001_R1_001.fastq.gz,/home/user/dragen_bcl_out/1_S1_L001_R2_001.fastq.gz
AATCCGTC.ACTGCATA.1,2,UnknownLibrary,1,/home/user/dragen_bcl_out/2_S2_
L001_R1_001.fastq.gz,/home/user/dragen_bcl_out/2_S2_L001_R2_001.fastq.gz
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
246
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

CGAACTTA.GCGTAAGA.1,3,UnknownLibrary,1,/home/user/dragen_bcl_out/3_S3_
L001_R1_001.fastq.gz,/home/user/dragen_bcl_out/3_S3_L001_R2_001.fastq.gz
GATAGACA.GCGTAAGA.1,4,UnknownLibrary,1,/home/user/dragen_bcl_out/4_S4_
L001_R1_001.fastq.gz,/home/user/dragen_bcl_out/4_S4_L001_R2_001.fastq.gz
Monitoring System Health
When you power up your DRAGEN system a daemon (dragen_mond) is started that monitors the card
for hardware issues.This daemon is also started when your DRAGEN system is installed or updated.
The main purpose of this daemon is to monitor DRAGEN Bio-IT Processor temperature and abort
DRAGEN when the temperature exceeds a configured threshold.
To manually start, stop, or restart the monitor, run the following as root:
sudo service dragen_mond [stop|start|restart]
By default, the monitor polls for hardware issues once per minute and logs temperature once every
hour.
The /etc/sysconfig/dragen_mond file specifies the command line options used to start dragen_
mond when the service command is run.Edit DRAGEN_MOND_OPTS in this file to change the default
options. For example, the following changes the poll time to 30 seconds and the log time to once every
2 hours:
DRAGEN_MOND_OPTS="-d -p 30 -l 7200"
The -d option is required to run the monitor as a daemon.
The dragen_mond command line options are as follows:
Option Description
-m --
swmaxtemp
<n>
Maximum software alarm temperature
(Celsius). Default is 85.
-i --
swmintemp
<n>
Minimum software alarm temperature
(Celsius). Default is 75.
-H --
hwmaxtemp
<n>
Maximum hardware alarm temperature
(Celsius). Default is 100.
-p --
polltime
<n>
Time between polling chip status register
(seconds). Default is 60.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
247
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Description
-l --logtime
<n>
Log FPGA temp every n seconds. Default
is 3600.Must be a multiple of polltime
-d --
daemon
Detach and run as a daemon.
-h --help Print help and exit.
-V --version Print the version and exit.
To display the current temperature of the DRAGEN Bio-IT Processor, use the dragen_info -t
command.This command does not execute if dragen_mond is not running.
% dragen_info -t
FPGA Temperature: 42C (Max Temp: 49C, Min Temp: 39C)
Logging
All hardware events are logged to /var/log/messages and /var/log/dragen_mond.log.The
following shows an example in /var/log/messages of a temperature alarm:
Jul 16 12:02:34 komodo dragen_mond[26956]: WARNING: FPGA software over temperature alarm has been triggered
-- temp threshold: 85 (Chip status: 0x80000001)
Jul 16 12:02:34 komodo dragen_mond[26956]: Current FPGA temp: 86, Max temp: 88, Min temp: 48
Jul 16 12:02:34 komodo dragen_mond[26956]: All dragen processes will be stopped until alarm clears
Jul 16 12:02:34 komodo dragen_mond[26956]: Terminating dragen in process 1510 with SIGUSR2 signal
By default, temperature is logged to /var/log/dragen_mond.log every hour:
Aug 01 09:16:50 Setting FPGA hardware max temperature threshold to 100
Aug 01 09:16:50 Setting FPGA software max temperature threshold to 85
Aug 01 09:16:50 Setting FPGA software min temperature threshold to 75
Aug 01 09:16:50 FPGA temperatures will be logged every 3600 seconds
Aug 01 09:16:50 Current FPGA temperature is 52 (Max temp = 52, Min temp = 52)
Aug 01 10:16:50 Current FPGA temperature is 53 (Max temp = 56, Min temp = 49)
Aug 01 11:16:50 Current FPGA temperature is 54 (Max temp = 56, Min temp = 49)
If DRAGEN is executing when a thermal alarm is detected, the following is displayed in the terminal
window of the DRAGEN process:
**********************************************************
** Received external signal -- aborting dragen. **
** An issue has been detected with the dragen card. **
** Check /var/log/messages for details. **
** **
** It may take up to a minute to complete shutdown. **
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
248
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
**********************************************************
If you see this message, stop running the DRAGEN software. Do the following to alleviate the
overheating condition on the card:
• Be sure that there is ample air flow over the card. Consider moving the card to a slot where there is
more air flow, adding another fan or increasing the fan speed.
• Give the card more space in the box.If there are available PCIe slots, move the card so that it has
empty slots on either side.
Contact Illumina Technical Support if you are having trouble resolving the thermal alarm on your
system.
Hardware Alarms
The following table lists the hardware events logged by the monitor when an alarm is triggered:
ID Description Monitor Action
0 Software
overheating
Terminate usage until DRAGEN Bio-IT Processor cools to software minimum
temperature.
1 Hardware
overheating
Fatal. Aborts dragen software; system reboot required
2 Board SPD
overheating
Logged as nonfatal
3 SODIMM
overheating
Logged as nonfatal
4 Power 0 Fatal. Aborts dragen software; system reboot required
5 Power 1 Fatal. Aborts dragen software; system reboot required
6 DRAGEN
Bio-IT
Processor
power
Logged as nonfatal
7 Fan 0 Logged as nonfatal
8 Fan 1 Logged as nonfatal
9 SE5338 Fatal. Aborts dragen software; system reboot required
10–
30
Undefined
(Reserved)
Fatal. Aborts dragen software; system reboot required
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
249
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Fatal alarms prevent the DRAGEN host software from running and require a system reboot.When a
software overheating alarm is triggered, the monitor looks for and aborts any running DRAGEN
processes.The monitor continues to abort any new DRAGEN processes until the temperature
decreases to the minimum threshold and the hardware clears the chip status alarm.When the
software overheating alarm clears, DRAGEN jobs can resume executing.
Contact Illumina Technical Support with details from the log files if any of these alarms are triggered on
your system.
Nirvana (Variant Annotator)
Nirvana provides clinical-grade annotation of genomic variants, such as SNVs, MNVs, insertions,
deletions, indels, STRs, SV, and CNVs. Use a VCF as input. The output is a structured JSON
representation of all annotations and sample information extracted from the VCF. Nirvana can handle
multiple alternate alleles and multiple samples.
DRAGEN provides a standalone implementation of the variant annotation software.
The following steps are required to run Nirvana.
1. Download external data sources, gene models, and reference genome.
2. Annotate the resulting JSON file.
By default, the Nirvana binaries are located in the /opt/edico/share/nirvana directory. This
directory includes two files: the Downloader and Nirvana.
Limitations
The Nirvana Variant Annotator and the Downloader are compatible with the following platforms:
• CentOS 6 using x64 processors.
• CentOS 7 and other modern Linux distributions using x64 processors.
Download Data Files
To store annotation data files, create a top-level directory. The created directory contains three
subdirectories:
• Cache contains gene models.
• SupplementaryAnnotation contains external data sources like dbSNP and gnomAD.
• References contains the reference genome.
The following command-line options are used:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
250
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Value Example Description
--ga GRCh37,
GRCh38,
or both
GRCh38 Genome assembly
--out Output
directory
~/Data Top-level output directory
Download data files as follows.
1. To create a data directory, enter the following command.
This example creates the Data directory in your home directory.
mkdir ~/Data
2. Download the files for a genome assembly.
This example downloads the genome assembly GRCh38.
/opt/edico/share/nirvana/Downloader --ga GRCh38 --out ~/Data
You can use the same command to resynchronize the data sources with the Nirvana servers, including
the following actions:
• Remove obsolete files, such as old versions of data sources, from the output directory.
• Download newer files.
The following is the created output:
---------------------------------------------------------------------------
Downloader (c) 2020 Illumina, Inc.
Stromberg, Roy, Lajugie, Jiang, Li, and Kang 3.9.1-0-gc823805
---------------------------------------------------------------------------
- downloading manifest... 37 files.
- downloading file metadata:
- finished (00:00:00.8).
- downloading files (22.123 GB):
- downloading 1000_Genomes_Project_Phase_3_v3_plus_refMinor.rma.idx
(GRCh38)
- downloading MITOMAP_20200224.nsa.idx (GRCh38)
- downloading ClinVar_20200302.nsa.idx (GRCh38)
- downloading REVEL_20160603.nsa.idx (GRCh38)
- downloading phyloP_hg38.npd.idx (GRCh38)
- downloading ClinGen_Dosage_Sensitivity_Map_20200131.nsi (GRCh38)
- downloading MITOMAP_SV_20200224.nsi (GRCh38)
- downloading dbSNP_151_globalMinor.nsa.idx (GRCh38)
- downloading ClinGen_Dosage_Sensitivity_Map_20190507.nga (GRCh38)
- downloading PrimateAI_0.2.nsa.idx (GRCh38)
- downloading ClinGen_disease_validity_curations_20191202.nga (GRCh38)
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
251
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
- downloading 1000_Genomes_Project_Phase_3_v3_plus.nsa.idx (GRCh38)
- downloading SpliceAi_1.3.nsa.idx (GRCh38)
- downloading dbSNP_153.nsa.idx (GRCh38)
- downloading TOPMed_freeze_5.nsa.idx (GRCh38)
- downloading MITOMAP_20200224.nsa (GRCh38)
- downloading gnomAD_2.1.nsa.idx (GRCh38)
- downloading ClinGen_20160414.nsi (GRCh38)
- downloading gnomAD_gene_scores_2.1.nga (GRCh38)
- downloading 1000_Genomes_Project_(SV)_Phase_3_v5a.nsi (GRCh38)
- downloading MultiZ100Way_20171006.pcs (GRCh38)
- downloading 1000_Genomes_Project_Phase_3_v3_plus_refMinor.rma (GRCh38)
- downloading ClinVar_20200302.nsa (GRCh38)
- downloading OMIM_20200409.nga (GRCh38)
- downloading Both.transcripts.ndb (GRCh38)
- downloading REVEL_20160603.nsa (GRCh38)
- downloading PrimateAI_0.2.nsa (GRCh38)
- downloading dbSNP_151_globalMinor.nsa (GRCh38)
- downloading Both.sift.ndb (GRCh38)
- downloading Both.polyphen.ndb (GRCh38)
- downloading Homo_sapiens.GRCh38.Nirvana.dat
- downloading 1000_Genomes_Project_Phase_3_v3_plus.nsa (GRCh38)
- downloading phyloP_hg38.npd (GRCh38)
- downloading SpliceAi_1.3.nsa (GRCh38)
- downloading TOPMed_freeze_5.nsa (GRCh38)
- downloading dbSNP_153.nsa (GRCh38)
- downloading gnomAD_2.1.nsa (GRCh38)
- finished (00:04:10.1).
Description Status
---------------------------------------------------------------------------
1000_Genomes_Project_(SV)_Phase_3_v5a.nsi (GRCh38) OK
1000_Genomes_Project_Phase_3_v3_plus.nsa (GRCh38) OK
1000_Genomes_Project_Phase_3_v3_plus.nsa.idx (GRCh38) OK
1000_Genomes_Project_Phase_3_v3_plus_refMinor.rma (GRCh38) OK
1000_Genomes_Project_Phase_3_v3_plus_refMinor.rma.idx (... OK
Both.polyphen.ndb (GRCh38) OK
Both.sift.ndb (GRCh38) OK
Both.transcripts.ndb (GRCh38) OK
ClinGen_20160414.nsi (GRCh38) OK
ClinGen_Dosage_Sensitivity_Map_20190507.nga (GRCh38) OK
ClinGen_Dosage_Sensitivity_Map_20200131.nsi (GRCh38) OK
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
252
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
ClinGen_disease_validity_curations_20191202.nga (GRCh38) OK
ClinVar_20200302.nsa (GRCh38) OK
ClinVar_20200302.nsa.idx (GRCh38) OK
Homo_sapiens.GRCh38.Nirvana.dat OK
MITOMAP_20200224.nsa (GRCh38) OK
MITOMAP_20200224.nsa.idx (GRCh38) OK
MITOMAP_SV_20200224.nsi (GRCh38) OK
MultiZ100Way_20171006.pcs (GRCh38) OK
OMIM_20200409.nga (GRCh38) OK
PrimateAI_0.2.nsa (GRCh38) OK
PrimateAI_0.2.nsa.idx (GRCh38) OK
REVEL_20160603.nsa (GRCh38) OK
REVEL_20160603.nsa.idx (GRCh38) OK
SpliceAi_1.3.nsa (GRCh38) OK
SpliceAi_1.3.nsa.idx (GRCh38) OK
TOPMed_freeze_5.nsa (GRCh38) OK
TOPMed_freeze_5.nsa.idx (GRCh38) OK
dbSNP_151_globalMinor.nsa (GRCh38) OK
dbSNP_151_globalMinor.nsa.idx (GRCh38) OK
dbSNP_153.nsa (GRCh38) OK
dbSNP_153.nsa.idx (GRCh38) OK
gnomAD_2.1.nsa (GRCh38) OK
gnomAD_2.1.nsa.idx (GRCh38) OK
gnomAD_gene_scores_2.1.nga (GRCh38) OK
phyloP_hg38.npd (GRCh38) OK
phyloP_hg38.npd.idx (GRCh38) OK
---------------------------------------------------------------------------
Peak memory usage: 52.3 MB
Time: 00:04:12.2
Annotate Files
1. If you have not generated a VCF file, download a VCF file using the following command
curl -O
https://raw.githubusercontent.com/HelixGrind/DotNetMisc/master/TestFiles/H
iSeq.10000.vcf.gz
2. To annotate the file, enter the following command:
/opt/edico/share/nirvana/Nirvana -c ~/Data/Cache/GRCh38/Both \ -r
~/Data/References/Homo_sapiens.GRCh38.Nirvana.dat \ --sd
~/Data/SupplementaryAnnotation/GRCh38 -i HiSeq.10000.vcf.gz -o HiSeq.10000
The following are the available command line options:
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
253
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Option Value Example Description
-c Directory ~/Data/Cache/GRCh38/Both Cache directory
-r Directory ~/Data/References/Homo_
sapiens.GRCh38.Nirvana.dat
Reference
directory
--sd Directory ~/Data/SupplementaryAnnotation/GRCh38 Supplementary
annotation
directory
-i path HiSeq.10000.vcf.gz Input VCF path
-o prefix HiSeq.10000 Output path prefix
Using the example above, Nirvana generates the following output called HiSeq.10000.json.gz.
---------------------------------------------------------------------------
Nirvana (c) 2020 Illumina, Inc.
Stromberg, Roy, Lajugie, Jiang, Li, and Kang 3.9.1-0-gc823805
---------------------------------------------------------------------------
Initialization Time Positions/s
---------------------------------------------------------------------------
Cache 00:00:01.9
SA Position Scan 00:00:00.4 23,867
Reference Preload Annotation Variants/s
---------------------------------------------------------------------------
chr1 00:00:00.4 00:00:03.7 2,651
Summary Time Percent
---------------------------------------------------------------------------
Initialization 00:00:02.3 25.7 %
Preload 00:00:00.4 5.4 %
Annotation 00:00:03.7 41.5 %
Peak memory usage: 1.284 GB
Time: 00:00:08.0
JSON Output file
Nirvana produces an output file in JSON format that includes the following three sections:
Section Content
Header Configuration, data source versions, and sample
names.
Positions Variant level annotation.
Genes Gene level annotation.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
254
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
The following are outputs using the example commands specified in the sections above. Each of the
following sections contains only a subset of information. The output file contains more information.
Header
The following is an example of the Header section.
"header": {
"annotator": "Nirvana 3.9.0",
"creationTime": "2020-06-03 08:05:06",
"genomeAssembly": "GRCh38",
"schemaVersion": 6,
"dataVersion": "91.26.57",
"dataSources": [
{
"name": "VEP",
"version": "91",
"description": "BothRefSeqAndEnsembl",
"releaseDate": "2018-03-05"
},
{
"name": "ClinVar",
"version": "20200302",
"description": "A freely accessible, public archive of reports of the
relationships among human variations and phenotypes, with supporting
evidence",
"releaseDate": "2020-03-02"
},
{
"name": "dbSNP",
"version": "153",
"description": "Identifiers for observed variants",
"releaseDate": "2019-07-22"
},
{
"name": "gnomAD",
"version": "2.1",
"description": "gnomAD allele frequency data remapped to GRCh38 with
CrossMap by Ensembl",
"releaseDate": "2019-03-25"
},
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
255
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
{
"name": "PrimateAI",
"version": "0.2",
"description": "PrimateAI percentile scores.",
"releaseDate": "2018-11-07"
},
{
"name": "OMIM",
"version": "20200409",
"description": "An Online Catalog of Human Genes and Genetic Disorders",
"releaseDate": "2020-04-09"
}
],
"samples": [
"NA12878"
]
},
Positions
Each position represents one row of the VCF file. Each position contains a samples section and a
variants section. Reference and alternate alleles shown here matches the VCF content exactly.
The samples section lists the sample-specific information, such as genotype, in the same order as they
appear in the VCF and in the JSON header above.
The variants section provides annotations for each alternate allele that appear in the VCF row. This
includes allele-specific annotation from external data sources as well as transcript-level annotation.
Reference and alternate alleles shown here are shown in their most shortened representation. For
example, padding bases have been removed and the variant are left-aligned.
"positions": [
{
"chromosome": "chr1",
"position": 1043248,
"refAllele": "C",
"altAlleles": [
"T"
],
"quality": 441.42,
"filters": [
"PASS"
],
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
256
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"strandBias": -425.94,
"cytogeneticBand": "1p36.33",
"samples": [
{
"genotype": "0/1",
"variantFrequencies": [
0.537
],
"totalDepth": 54,
"genotypeQuality": 99,
"alleleDepths": [
25,
29
]
}
],
"variants": [
{
"vid": "1-1043248-C-T",
"chromosome": "chr1",
"begin": 1043248,
"end": 1043248,
"refAllele": "C",
"altAllele": "T",
"variantType": "SNV",
"hgvsg": "NC_000001.11:g.1043248C>T",
"phylopScore": 0.1,
"clinvar": [
{
"id": "RCV000872112.1",
"variationId": 263161,
"reviewStatus": "criteria provided, single submitter",
"alleleOrigins": [
"germline"
],
"refAllele": "C",
"altAllele": "T",
"phenotypes": [
"not provided"
],
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
257
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"medGenIds": [
"CN517202"
],
"significance": [
"likely benign"
],
"lastUpdatedDate": "2019-12-17",
"pubMedIds": [
"28492532"
],
"isAlleleSpecific": true
},
{
"id": "VCV000263161.2",
"reviewStatus": "criteria provided, multiple submitters, no conflicts",
"significance": [
"likely benign"
],
"refAllele": "C",
"altAllele": "T",
"lastUpdatedDate": "2019-12-17",
"isAlleleSpecific": true
}
],
"dbsnp": [
"rs116586548"
],
"globalAllele": {
"globalMinorAllele": "T",
"globalMinorAlleleFrequency": 0.004393
},
"gnomad": {
"coverage": 38,
"allAf": 0.000681,
"allAn": 264462,
"allAc": 180,
"allHc": 0,
"afrAf": 0.006216,
"afrAn": 23648,
"afrAc": 147,
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
258
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"afrHc": 0,
"amrAf": 0.000689,
"amrAn": 33404,
"amrAc": 23,
"amrHc": 0,
"easAf": 0,
"easAn": 18830,
"easAc": 0,
"easHc": 0,
"finAf": 0,
"finAn": 22870,
"finAc": 0,
"finHc": 0,
"nfeAf": 5e-05,
"nfeAn": 120576,
"nfeAc": 6,
"nfeHc": 0,
"asjAf": 0.000304,
"asjAn": 9882,
"asjAc": 3,
"asjHc": 0,
"sasAf": 0,
"sasAn": 28456,
"sasAc": 0,
"sasHc": 0,
"othAf": 0.000147,
"othAn": 6796,
"othAc": 1,
"othHc": 0,
"maleAf": 0.000564,
"maleAn": 143614,
"maleAc": 81,
"maleHc": 0,
"femaleAf": 0.000819,
"femaleAn": 120848,
"femaleAc": 99,
"femaleHc": 0,
"controlsAllAf": 0.000626,
"controlsAllAn": 113456,
"controlsAllAc": 71
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
259
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
},
"oneKg": {
"allAf": 0.004393,
"afrAf": 0.016641,
"amrAf": 0,
"easAf": 0,
"eurAf": 0,
"sasAf": 0,
"allAn": 5008,
"afrAn": 1322,
"amrAn": 694,
"easAn": 1008,
"eurAn": 1006,
"sasAn": 978,
"allAc": 22,
"afrAc": 22,
"amrAc": 0,
"easAc": 0,
"eurAc": 0,
"sasAc": 0
},
"primateAI": [
{
"hgnc": "AGRN",
"scorePercentile": 0.12
}
],
"revel": {
"score": 0.136
},
"spliceAI": [
{
"hgnc": "AGRN",
"acceptorGainScore": 0.1,
"acceptorGainDistance": 23,
"acceptorLossScore": 0,
"acceptorLossDistance": -9,
"donorGainScore": 0,
"donorGainDistance": -5,
"donorLossScore": 0,
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
260
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"donorLossDistance": 16
}
],
"topmed": {
"allAf": 0.002055,
"allAn": 125568,
"allAc": 258,
"allHc": 1
},
"transcripts": [
{
"transcript": "ENST00000379370.6",
"source": "Ensembl",
"bioType": "protein_coding",
"codons": "cCg/cTg",
"aminoAcids": "P/L",
"cdnaPos": "1444",
"cdsPos": "1394",
"exons": "8/36",
"proteinPos": "465",
"geneId": "ENSG00000188157",
"hgnc": "AGRN",
"consequence": [
"missense_variant"
],
"hgvsc": "ENST00000379370.6:c.1394C>T",
"hgvsp": "ENSP00000368678.2:p.(Pro465Leu)",
"isCanonical": true,
"polyPhenScore": 0.065,
"polyPhenPrediction": "benign",
"proteinId": "ENSP00000368678.2",
"siftScore": 0.05,
"siftPrediction": "tolerated"
},
{
"transcript": "NM_198576.3",
"source": "RefSeq",
"bioType": "protein_coding",
"codons": "cCg/cTg",
"aminoAcids": "P/L",
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
261
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"cdnaPos": "1444",
"cdsPos": "1394",
"exons": "8/36",
"proteinPos": "465",
"geneId": "375790",
"hgnc": "AGRN",
"consequence": [
"missense_variant"
],
"hgvsc": "NM_198576.3:c.1394C>T",
"hgvsp": "NP_940978.2:p.(Pro465Leu)",
"isCanonical": true,
"polyPhenScore": 0.065,
"polyPhenPrediction": "benign",
"proteinId": "NP_940978.2",
"siftScore": 0.05,
"siftPrediction": "tolerated"
}
]
}
]
}
],
Genes
For each gene referenced in the transcripts in the positions section, there is a matching entry in the
genes section. The following example shows gene-level annotations from gnomAD, ClinGen Dosage
Sensitivity Map, and OMIM.
"genes": [
{
"name": "AGRN",
"gnomAD": {
"pLi": 5.47e-07,
"pRec": 1,
"pNull": 1.41e-12,
"synZ": -3.96,
"misZ": 0.226,
"loeuf": 0.435
},
"clingenDosageSensitivityMap": {
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
262
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
"haploinsufficiency": "gene associated with autosomal recessive phenotype",
"triplosensitivity": "no evidence to suggest that dosage sensitivity is
associated with clinical phenotype"
},
"omim": [
{
"mimNumber": 103320,
"geneName": "Agrin",
"description": "The AGRN gene encodes agrin, a large and ubiquitous
proteoglycan with multiple isoforms that have diverse functions in
different tissues. Agrin was originally identified as an essential neural
regulator that induces the aggregation of acetylcholine receptors (AChRs)
and other postsynaptic proteins on muscle fibers and is crucial for the
formation and maintenance of the neuromuscular junction (NMJ) (Campanelli
et al., 1991; Burgess et al., 1999; summary by Maselli et al., 2012).",
"phenotypes": [
{
"mimNumber": 615120,
"phenotype": "Myasthenic syndrome, congenital, 8, with pre- and
postsynaptic defects",
"description": "Congenital myasthenic syndromes are genetic disorders of
the neuromuscular junction (NMJ) that are classified by the site of the
transmission defect: presynaptic, synaptic, and postsynaptic. CMS8 is an
autosomal recessive disorder characterized by prominent defects of both the
pre- and postsynaptic regions. Affected individuals have onset of muscle
weakness in early childhood; the severity of the weakness and muscles
affected is variable (summary by Maselli et al., 2012).\n\nFor a discussion
of genetic heterogeneity of CMS, see CMS1A.",
"mapping": "molecular basis of the disorder is known",
"inheritances": [
"Autosomal recessive"
]
}
]
}
]
}
]
}
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
263
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Hardware-Accelerated Compression and Decompression
Gzip compression is ubiquitous in bioinformatics. FASTQ files are often gzipped, and the BAM format
itself is a specialized version of gzip. For that reason, the DRAGEN BioIT processor provides hardware
support for accelerating compression and decompression of gzipped data.If your input files are
gzipped, DRAGEN detects that and decompresses the files automatically. If your output is BAM files,
then the files are automatically compressed.
DRAGEN provides standalone command-line utilities to enable you to compress or decompress
arbitrary files.These utilities are analogous to the Linux gzip and gunzip commands, but are named
dzip and dunzip (dragen zip and dragen unzip).Both utilities are able to accept as input a single file, and
produce a single output file with the .gz file extension removed or added, as appropriate. For example:
dzip file1 # produces output file file1.gz
dunzip file2.gz # produces output file file2
Currently, dzip and dunzip have the following limitations and differences from gzip/gunzip:
• Each invocation of these tools can handle only a single file.Additional file names (including those
produced by a wildcard * character) are ignored.
• They cannot be run at the same time as the DRAGEN host software.
• They do not support the command line options found in gzip and gunzip (eg, --recursive, --fast,
--best, --stdout).
Usage Reporting
As part of the install process, a daemon (dragen_licd) is created (or stopped then restarted). This
background process self-activates at the end of each day to upload DRAGEN host software usage to
anIllumina server. Information includes date, duration, size (number of bases), status of each run, and
software version used.
Communication to theIllumina server is secured by encryption. If there is a communication error, the
daemon retries until the next morning. If the upload continues to fail, communication is tried again the
following night until communication succeeds. This means that during working hours, the system
resources remain fully available and are not in any way hampered by this background activity.
To check the current license usage, use the dragen_lic command.
To generate a usage report, your server must meet the following requirements:
• 256 GB RAM and 2 TB HDD for 300x germline single sample coverage.
• T/N analysis coverage.
• 6 TB and 512 GB RAM.
The usage report does not contain the following information:
• Number of joint genotyper input files.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
264
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
• GATK gVCF input to gVCF genotyper.
• Mixing gVCFs from different callers, such as joint calling and gVCF genotyper.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
265
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
Troubleshooting
If the DRAGEN system does not seem to be responding, do the following:
1. To determine if the DRAGEN system is hanging, follow the instructions in How to Determine if the
System is Hanging.
2. Collect diagnostic information after a hang, or a crash, as described in Sending Diagnostic
Information to Illumina Support.
3. After all information has been collected, reset your system. if needed, as described in Resetting
Your System after a Crash or Hang.
How to Determine if the System is Hanging
The DRAGEN system has a watchdog to monitor the system for hangs. If a run seems to be taking
longer than it should, the watchdog may not be detecting the hang.Here are some things to try:
• Run the top command to find the active DRAGEN process.If your run is healthy, you should expect
to see it consuming over 100% of the CPU.If it is consuming 100% or less, then your system may be
hanging.
• Run the du -s command in the directory of the output BAM/SAM file.During a normal run, this
directory should be growing with either intermediate output data (when sort is enabled) or
BAM/SAM data.
Sending Diagnostic Information to Illumina Support
Illumina would like your feedback on your DRAGEN system, including any reports of system
malfunction.In the event of a crash, hang, or watchdog fault, run the sosreport command to collect
diagnostic and configuration information, as follows:
sudo sosreport --batch --tmp-dir /staging/tmp
This command takes several minutes to execute and reports the location where it has saved the
diagnostic information in /staging/tmp.Please include the report when you submit a support ticket
for Illumina Technical Support.
Resetting Your System after a Crash or Hang
If the DRAGEN system crashes or hangs, the dragen_reset utility must be run to reinitialize the
hardware and software.This utility is automatically executed by the host software any time it detects
an unexpected condition.In this case, the host software shows the following message:
Running dragen_reset to reset DRAGEN Bio-IT processor and software
If the software is hanging, please collect diagnostic information as described in subsection Sending
Diagnostic Information to Illumina Support on page 266 and then execute dragen_reset manually, as
follows:
/opt/edico/bin/dragen_reset
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
266
Any execution of dragen_reset requires the reference genome to be reloaded to the DRAGEN board.
The host software automatically reloads the reference on the next execution.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
267
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Command Line Option Reference
This section provides information on all the DRAGEN command line options, including the name used in
the configuration file, the command line equivalent, a description, and the range of values.
General Software Options
The following options are in the default section of the configuration file.The default section does not
have a section name (eg, [Aligner]) associated with it. The default section is at the top of the
configuration file. Note that some mandatory fields must be specified on the command line and are not
present in configuration files.
Name Description
Command Line
Equivalent
Value
alt-aware Enables special processing for
alt contigs, if alt liftover was
used in hash table.Enabled by
default if reference was built
with liftover.
--alt-aware true/false
append-read-index-
to-name
By default, DRAGEN names both
mate ends of pairs the
same.When set to true,
DRAGEN appends /1 and /2 to
the two ends.
true/false
bam-input Aligned BAM file for input to the
DRAGEN variant caller.
-b, --bam-input
bcl-conversion-only Perform Illumina BCL conversion
to FASTQ format.
--bcl-conversion-only true/false
bcl-input-directory Input BCL directory for BCL
conversion.
--bcl-input-directory
bcl-only-lane For BCL input, convert only
specified lane number (default
all lanes).
--bcl-only-lane 1–8
sample-sheet For BCL input, path to
SampleSheet.csv file. Default
location is the BCL root
directory.
--sample-sheet
strict-mode For BCL input, abort if any files
are missing (false by default)
--strict-mode true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
268

Name Description
Command Line
Equivalent
Value
first-tile-only Only convert the first tile of each
lane during BCL conversion (for
testing/debugging)
--first-tile-only true/false
run-info Set the path to RunInfo.xml
file. Default is <flow
cell>/RunInfo.xml
--run-info
bcl-sampleproject-
subdirectories
For BCL conversion, output to
subdirectories based upon
sample sheet 'Sample_Project'
column
--bcl-sampleproject-
subdirectories
no-lane-splitting Set whether to output all lanes
of a flow cell to the same FASTQ
files consecutively. Default is
false.
--no-lane-splitting true/false
bcl-only-matched-
reads
Set whether to output
unmapped reads to files marked
as Undetermined. Default is
false.
bcl-only-matched-
reads
true/false
bcl-use-hw Set to false to prevent use of
DRAGENFPGA acceleration
during BCL conversion. Default
is true.
--bcl-use-hw true/false
bcl-num-parallel-tiles Number of tiles to process in
parallel. Default is dynamically
determined.
--bcl-num-parallel-
tiles
1-<nproc>
bcl-num-conversion-
threads
Number of conversion threads
per tile. Default is dynamically
determined.
--bcl-num-
conversion-threads
1-<nproc>
bcl-num-
compression-threads
Number of CPU threads for
output fastq.gz compression.
Default is dynamically
determined.
--bcl-num-
compression-threads
1-<nproc>
bcl-num-
decompression-threads
Number of CPU threads for BCL
input decompression. Default is
dynamically determined.
--bcl-num-
decompression-threads
1-<nproc>
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
269
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
shared-thread-
odirect-output
Use alternative shared-thread
ODIRECT file output. Default is
false.
--shared-thread-
odirect-output
true/false
build-hash-table Generate a reference/hash
table.
--build-hash-table true/false
cram-input CRAM file for input to the
DRAGENvariant caller.
--cram-input
dbsnp Path to the variant annotation
database VCF (or .vcf.gz) file.
--dbsnp
enable-auto-multifile Import subsequent segments of
the *_001.{dbam,fastq} files.
--enable-auto-
multifile
true/false
enable-bam-indexing Enable generation of a BAI index
file.
--enable-bam-
indexing
true/false
enable-cnv Enable copy number variant
(CNV).
--enable-cnv true/false
enable-duplicate-
marking
Enable the flagging of duplicate
output alignment records.
--enable-duplicate-
marking
true/false
enable-map-align-
output
Enables saving the output from
the map/align stage. Default is
true when only running
map/align. Default is false if
running the variant caller.
--enable-map-align-
output
true/false
enable-methylation-
calling
Whether to automatically add
methylation-tags and output a
single BAM for methylation
protocols.
true/false
enable-sampling Automatically detect paired-end
parameters by running a sample
through the mapper/aligner.
true/false
enable-sort Enable sorting after
mapping/alignment.
true/false
enable-variant-caller Enables the variant caller. --enable-variant-
caller
true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
270
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
enable-vcf-
compression
Enable compression of VCF
output files. Default is true.
true/false
fastq-file1 FASTQ file to input to the
DRAGEN pipeline (may be
gzipped).
-1, --fastq-file1
fastq-file2 Second FASTQ file with paired-
end reads for input.
-2, --fastq-file2
fastq-list CSV file that contains a list of
FASTQ files to process.
--fastq-list
fastq-list-sample-id Only process entries where the
RGSM entry matches the given
Sample ID parameter (for fastq-
list.csv input).
--fastq-list-sample-id true/false
fastq-list-all-samples Enable/disable processing of all
samples together, regardless of
the RGSMvalue.
--fastq-list-all-
samples
true/false
fastq-n-quality Base call quality to output for N
bases.Automatically added to
fastq-n-quality for all output N’s.
--fastq-n-quality 0 to 255
fastq-offset FASTQ quality offset value. --fastq-offset 33 or 64
filter-flags-from-
output
Filter output alignments with any
bits set in val present in the flags
field. Hex and decimal values
accepted.
--filter-flags-from-
output
force Force overwrite of existing
output file.
-f
force-load-reference Force loading of the reference
and hash tables before starting
the DRAGEN pipeline.
-l
generate-md-tags Whether to generate MD tags
with alignment output records.
Default is false.
--generate-md-tags true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
271
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
generate-sa-tags Whether to generate SA:Z tags
for records that have
chimeric/supplemental
alignments.
--generate-sa-tags true/false
generate-zs-tags Whether to generate ZS tags for
alignment output
records.Default is false.
--generate-sz-tags true/false
ht-alt-liftover SAM format liftover file of
alternate contigs in reference.
--ht-alt-liftover
ht-pop-alt-contigs Reference FASTA file with
population alternate contigs to
augment the standard
reference.
--ht-pop-alt-contigs
ht-pop-alt-liftover SAM format liftover file of
population alternate contigs.
--ht-pop-alt-liftover
ht-pop-snps VCF format file with unphased
population SNPs used to
augment the standard
reference.
--ht-pop-snps
ht-alt-aware-validate Disable requirement for a liftover
file when building a hash table
from a reference that contains
alt-contigs.
--ht-alt-aware-
validate
true/false
ht-build-rna-
hashtable
Enable generation of RNA hash
table. Default is false.
--ht-build-rna-
hashtable
true/false
ht-cost-coeff-seed-
freq
Cost coefficient of extended
seed frequency.
--ht-cost-coeff-seed-
freq
ht-cost-coeff-seed-
len
Cost coefficient of extended
seed length.
--ht-cost-coeff-seed-
len
ht-cost-penalty-incr Cost penalty to incrementally
extend a seed another step.
--ht-cost-penalty-incr
ht-cost-penalty Cost penalty to extend a seed by
any number of bases.
--ht-cost-penalty
ht-decoys Specifies the path to a decoys
file.
--ht-decoys
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
272
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
ht-max-dec-factor Maximum decimation factor for
seed thinning.
--ht-max-dec-factor
ht-max-ext-incr Maximum bases to extend a
seed by in one step.
--ht-max-ext-incr
ht-max-ext-seed-len Maximum extended seed length. -- ht-max-ext-seed-
len
ht-max-seed-freq Maximum allowed frequency for
a seed match after extension
attempts.
--ht-max-seed-freq 1–256
ht-max-table-chunks Maximum ~1 GB thread table
chunks in memory at once.
--ht-max-table-
chunks
ht-mem-limit Memory limit (hash table +
reference), units B|KB|MB|GB.
--ht-mem-limit
ht-methylated Automatically generate C->T
and G->A converted reference
hashtables.
--ht-methylated true/false
ht-num-threads Maximum worker CPU threads
for building hash table.
--ht-num-threads
ht-rand-hit-extend Include a random hit with each
EXTEND record of this freq
record.
--ht-rand-hit-extend
ht-rand-hit-hifreq Include a random hit with each
HIFREQ record.
--ht-rand-hit-hifreq
ht-ref-seed-interval Number of positions per
reference seed.
--ht-ref-seed-interval
ht-reference Reference file in .fasta format to
be used to build a hash table.
--ht-reference
ht-seed-len Initial seed length to store in
hash table.
--ht-seed-len
ht-size Size of hash table, units
B|KB|MB|GB.
--ht-size
ht-soft-seed-freq-cap Soft seed frequency cap for
thinning
--ht-soft-seed-freq-
cap
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
273
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
ht-suppress-decoys Suppress the use of a decoys file
when building a hash table.
--ht-suppress-decoys
ht-target-seed-freq Target seed frequency for seed
extension.
--ht-target-seed-freq
input-qname-suffix-
delimiter
Controls the delimiter used for
append-read-index-to-name
and for detecting matching pair
names with BAM input.
/ or
. or
:
interleaved Interleaved paired-end reads in
single FASTQ.
-i
intermediate-results-
dir
Directory to store intermediate
results in (eg, sort partitions).
lic-no-print Suppress the license status
message at the end of a run.
--lic-no-print true/false
methylation-
generate-cytosine-
report
Generate a genomewide
cytosine methylation report.
--methylation-
generate-cytosine-
report
true/false
methylation-
generate-mbias-
report
Generate a per-sequencer-
cyclemethylation bias report.
true/false
methylation-TAPS If input assays are generated by
TAPS, set to true.
--methylation-TAPS true/false
methylation-match-
bismark
If true, match bismark’s tags
exactly, including bugs.
--methylation-match-
bismark
true/false
methylation-protocol Library protocol for methylation
analysis.
--methylation-
protocol
none /
directional /
nondirectional
/
directional-
complement
num-threads The number of processor
threads to use.
-n, --num-threads
output-directory Output directory. --output-directory
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
274
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
output-file-prefix Output file name prefix to use for
all files generated by the
pipeline.
--output-file-prefix
output-format The format of the output file
from the map/align stage.Valid
values are bam (the default),
sam, or dbam (a proprietary
binary format).
--output-format BAM / SAM /
DBAM
pair-by-name Whether to shuffle the order of
BAM input records such that
paired-end mates are processed
together.
pair-suffix-delimiter Change the delimiter character
for suffixes.
--pair-suffix-delimiter / . :
preserve-bqsr-tags Whether to preserve input BAM
file’s BI and BD flags.Note this
may cause problems with hard
clipping.
true/false
preserve-map-align-
order
Produce output file that
preserves original order of reads
in the input file.
true/false
qc-coverage-region-1 First bed file to report coverage
on.
--qc-coverage-
region-1
qc-coverage-region-2 Second bed file to report
coverage on.
--qc-coverage-
region-2
qc-coverage-region-3 Third bed file to report coverage
on.
--qc-coverage-
region-3
qc-coverage-reports-
1
Types of reports requested for
qc-coverage-region-1.
--qc-coverage-
reports-1
full_res / cov_
report
qc-coverage-reports-
2
Types of reports requested for
qc-coverage-region-2.
--qc-coverage-
reports-2
full_res / cov_
report
qc-coverage-reports-
3
Types of reports requested for
qc-coverage-region-3.
--qc-coverage-
reports-3
full_res / cov_
report
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
275
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
ref-dir Directory containing the
reference hash table. This
reference is automatically
loaded to the DRAGEN card, if it
is not already there.
-r, --ref-dir
ref-sequence-filter Output only reads mapping to
this reference sequence.
--ref-sequence-filter
remove-duplicates If true, remove duplicate
alignment records instead of just
flagging them.
true/false
RGCN Read group sequencing center
name.
--RGCN
RGCN-tumor Read group sequencing center
name for tumor input.
--RGCN-tumor
RGDS Read group description. --RGDS
RGDS-tumor Read group description for
tumor input.
--RGDS-tumor
RGDT Read group run date. --RGDT
RGDT-tumor Read group run date for tumor
input.
--RGDT-tumor
RGID Read group ID. --RGID
RGID-tumor Read group ID for tumor input. --RGID-tumor
RGLB Read group library. --RGLB
RGLB-tumor Read group library for tumor
input.
--RGLB-tumor
RGPI Read group predicted insert size. --RGPI
RGPI-tumor Read group predicted insert size
for tumor input.
--RGPI-tumor
RGPL Read group sequencing
technology.
--RGPL
RGPL-tumor Read group sequencing
technology for tumor input.
--RGPL-tumor
RGPU Read group platform unit. --RGPU
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
276
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
RGPU-tumor Read group platform unit for
tumor input.
--RGPU-tumor
RGSM Read group sample name. --RGSM
RGSM-tumor Read group sample name for
tumor input.
--RGSM-tumor
sample-size Number of reads to sample
when enable-sampling is true.
sample-sex Sex of the sample. --sample-sex
strip-input-qname-
suffixes
Whether to strip read-index
suffixes (eg, /1 and /2) from
input QNAMEs.
true/false
tumor-bam-input Aligned BAM file for the DRAGEN
variant caller in somatic mode.
--tumor-bam-input
tumor-cram-input Aligned CRAM file for the
DRAGEN variant caller in somatic
mode.
--tumor-cram-input
tumor-fastq-list A CSV file containing a list of
FASTQfiles for the mapper,
aligner, and somatic variant
caller.
--tumor-fastq-list
tumor-fastq-list-
sample-id
The sample ID for the list of
FASTQ files specified by tumor-
fastq-list.
--tumor-fastq-list-
sample-id
tumor-fastq1 FASTQ file for the DRAGEN
pipeline using the variant caller
in somatic mode (may be
gzipped).
--tumor-fastq1
tumor-fastq2 Second FASTQ file with reads
paired to tumor-fastq1 reads for
the DRAGEN pipeline using the
variant caller in somatic mode
(may be gzipped).
--tumor-fastq2
verbose Enable verbose output from
DRAGEN.
-v
version Print the version and exit. -V
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
277
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Mapper Options
The following options are in the [Mapper] section of the configuration file.For more detailed
information on these options, see DNA Mapping on page 62.
Name Description
Command Line
Equivalent
Range
ann-sj-
max-indel
Maximum indel length to expect
near an annotated splice junction.
--Mapper.ann-sj-
max-indel
0 to 63
edit-chain-
limit
For edit-mode 1 or 2: Maximum seed
chain length in a read to qualify for
seed editing.
--Mapper.edit-chain-
limit
edit-chain-limit
>= 0
edit-mode 0 = No edits, 1 = Chain len test, 2 =
Paired chain len test, 3 = Edit all std
seeds.
--Mapper.edit-mode 0 to 3
edit-read-
len
For edit-mode 1 or 2: Read length in
which to try edit-seed-num edited
seeds.
--Mapper.edit-read-
len
edit-read-len > 0
edit-seed-
num
For edit-mode 1 or 2: Requested
number of seeds per read to allow
editing on.
--Mapper.edit-seed-
num
edit-seed-num
>= 0
enable-
map-align
Enable use of BAMinput files for
mapper/aligner.
--enable-map-align true/false
map-
orientations
0=Normal, 1=No Rev Comp, 2=No
Forward (paired end requires
Normal).
--Mapper.map-
orientations
0 to 2
max-intron-
bases
Maximum intron length reported. --Mapper.max-
intron-bases
min-intron-
bases
Minimum reference deletion length
reported as an intron.
--Mapper.min-intron-
bases
seed-
density
Requested density of seeds from
reads queried in the hash table
--Mapper.seed-
density
0 > seed-density
> 1
Aligner Options
The following options are in the [Aligner] section of the configuration file.For more information, see
DNAAligning on page 65
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
278
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
aln-min-score (signed) Minimum
alignment score to report;
baseline for MAPQ.
When using local
alignments (global = 0), aln-
min-score is computed by
the host software as "22 *
match-score".
When using global
alignments (global = 1), aln-
min-score is set to -
1000000.
Host software computation
may be overridden by
setting aln-min-score in
configuration file.
--Aligner.aln-min-
score
−2,147,483,648
to
2,147,483,647
dedup-min-qual Minimum base quality for
calculating read quality
metric for deduplication.
--Aligner.dedup-min-
qual
0–63
en-alt-hap-aln Allows chimeric alignments
to be output, as
supplementary.
--Aligner.en-alt-hap-
aln
0–1
en-chimeric-aln Allows chimeric alignments
to be output, as
supplementary.
--Aligner.en-
chimeric-aln
0–1
gap-ext-pen Score penalty for gap
extension.
--Aligner.gap-ext-
pen
0–15
gap-open-pen Score penalty for opening a
gap (insertion or deletion).
gap-open-pen 0–127
global If alignment is global
(Needleman-Wunsch)
rather than local (Smith-
Waterman).
--Aligner.global 0–1
hard-clips Flags for hard clipping: [0]
primary, [1] supplementary,
[2] secondary.
--Aligner.hard-clips 3 bits
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
279
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
map-orientations Constrain orientations to
accept forward-only,
reverse-complement only,
or any alignments.
--Aligner.map-
orientations
0 (any)
1 (forward only)
2 (reverse only)
mapq-max Ceiling on reported MAPQ. --Aligner.mapq-max 0 to 255
mapq-strict-js Specific to RNA. When set
to 0, a higher MAPQ value is
returned, expressing
confidence that the
alignment is at least
partially correct. When set
to 1, a lower MAPQ value is
returned, expressing the
splice junction ambiguity.
--mapq-strict-js 0/1
match-n-score (signed) Score increment
for matching a reference 'N'
nucleotide IUB code.
--Aligner.match-n-
score
-16–15
match-score Score increment for
matching reference
nucleotide.
--Aligner.match-
score
When global =
0, match-score
> 0; When
global = 1,
match-score >=
0
max-rescues Maximum rescue
alignments per read pair.
Default is 10.
--max-rescues 0–1023
min-score-coeff Adjustment to aln-min-
score per read base.
--Aligner. min-score-
coeff
-64–63.999
mismatch-pen Score penalty for a
mismatch.
--Aligner.mismatch-
pen
0–63
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
280
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
no-unclip-score When no-unclip-score is set
to 1, any unclipped bonus
(unclip-score) contributing
to an alignment is removed
from the alignment score
before further processing.
--Aligner.no-unclip-
score
0-1
no-unpaired If only properly paired
alignments should be
reported for paired reads.
--Aligner. no-
unpaired
0–1
pe-max-penalty Maximum pairing score
penalty, for unpaired or
distant ends.
--Aligner.pe-max-
penalty
0–255
pe-orientation Expected paired-end
orientation: 0=FR, 1=RF,
2=FF.
--Aligner.pe-
orientation
0, 1, 2
rescue-sigmas Deviations from the mean
read length used for rescue
scan radius. Default is 2.5.
--Aligner.rescue-
sigmas
sec-aligns Maximum secondary
(suboptimal) alignments to
report per read.
--Aligner.sec-aligns 0–30
sec-aligns-hard Set to force unmapped
when not all secondary
alignments can be output.
--Aligner.sec-aligns-
hard
0–1
sec-phred-delta Only secondary alignments
with likelihood within this
Phred of the primary are
reported.
--Aligner.sec-phred-
delta
0–255
sec-score-delta Secondary aligns allowed
with pair score no more
than this far below primary.
--Aligner. sec-score-
delta
supp-aligns Maximum supplementary
(chimeric) alignments to
report per read.
--Aligner.supp-aligns 0–30
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
281
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
supp-as-sec If supplementary
alignments should be
reported with secondary
flag.
--Aligner.supp-as-
sec
0–1
supp-min-score-adj Amount to increase
minimum alignment score
for supplementary
alignments. This score is
computed by host software
as "8 * match-score" for
DNA, and is default 0 for
RNA.
--Aligner. supp-min-
score-adj
unclip-score Score bonus for reaching
each edge of the read.
--Aligner.unclip-
score
0–127
unpaired-pen Penalty for unpaired
alignments in Phred scale.
--Aligner.unpaired-
pen
0–255
If you disable automatic detection of insert-length statistics via the --enable-sampling option, you must
override all the following options to specify the statistics. For more information, see Mean Insert Size
Detection on page 1.These options are part of the [Aligner] section of the configuration file.
Option Description
Command Line
Equivalent
Value
pe-stat-mean-insert Average template length. 0–65535
pe-stat-mean-read-
len
Average read length. 0–65535
pe-stat-quartiles-
insert
A comma-delimited trio of
numbers specifying 25
th
, 50
th
,
and 75
th
percentile template
lengths.
0–65535
pe-stat-stddev-insert Standard deviation of template
length distribution.
0–65535
Variant Caller Options
The following options are in the Variant Caller section of the configuration file. For more information on
these options, see Variant Caller Options on page 83.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
282
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
dn-cnv-vcf Joint structural variant
VCF from the CNV
calling step. If omitted,
checks with overlapping
copy number variants
are skipped.
--dn-cnv-vcf
dn-input-vcf Joint small variant VCF
de novo calling step to
be filtered
--dn-input-vcf
dn-output-vcf File location to which the
filtered VCF should be
written. If not specified,
the input VCF is
overwritten.
--dn-output-vcf
dn-sv-vcf Joint structural variant
VCF from the SV calling
step. If omitted, checks
with overlapping
structural variants are
skipped.
--dn-sv-vcf
enable-
combinegvcfs
Enable/disable combine
gVCF run.
--enable-
combinegvcfs
true/false
enable-joint-
genotyping
To enable combine gVCF
run, set to true.
--enable-joint-
genotyping
true/false
enable-multi-
sample-gvcf
Enable/disable
generation of a
multisample gVCF file. If
set to true, requires a
combined gVCF file as
input.
--enable-multi-
sample-gvcf
true/false
enable-sv Enable/disable structural
variant caller. Default is
false.
--enable-sv true/false
enable-vlrd Enable/disable Virtual
Long Read Detection.
--enable-vlrd true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
283
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
panel-of-normals The path to the panel of
normals VCF file.
--panel-of-normals
pedigree-file Specific to joint calling.
The path to a PED
pedigree file containing a
structured description of
the familial relationships
between samples. The
pedigree file can contain
trios. Only pedigree files
that contain trios are
supported.
--pedigree-file
qc-snp-DeNovo-
quality-threshold
The threshold for
counting and reporting
De Novo SNP variants.
--qc-snp-DeNovo-
quality-threshold
qc-indel-DeNovo-
quality-threshold
The threshold for
counting and reporting
De Novo INDEL variants.
--qc-indel-DeNovo-
quality-threshold
variant The path to a single
gVCF file. Multiple --
variant options can be
used on the command
line, one
for each gVCF. Up to 500
gVCFs are supported.
--variant
variant-list The path to a file
containing a list of input
gVCF files, one file per
line, that need to be
combined.
--variant-list
vc-af-call-threshold Set the allele frequency
call threshold to emit a
call in the VCF if the AF
filter is enabled. The
default is 0.01.
--vc-af-call-
threshold
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
284
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-af-filter-threshold Set the allele frequency
filter threshold to mark
emitted VCF calls as
filtered if the AF filter is
enabled. The default is
0.05.
--vc-af-filter-
threshold
vc-callability-normal-
threshold
The normal sample
coverage threshold for a
site to be considered
callable in the somatic
callable regions report.
--vc-callability-
normal-thresh
vc-callability-tumor-
threshold
The tumor sample
coverage threshold for a
site to be considered
callable in the somatic
callable regions report.
--vc-callability-
tumor-thresh
vc-decoy-contigs The path to a comma-
separated list of contigs
to skip during variant
calling.
--vc-decoy-contigs
vc-detect-
systematic-noise
Sensitive VCF run mode
used when building a
systematic noise file.
--vc-detect-
systematic-noise
true/false
vc-emit-ref-
confidence
To enable base pair
gVCF generation, set to
BP_RESOLUTION. To
enable banded gVCF
generation, set to GVCF.
--vc-emit-ref-
confidence
BP_RESOLUTION /
GVCF
vc-enable-af-filter Enable/disable the allele
frequency filter for
somatic mode. Default is
false.
--vc-enable-af-filter true/false
vc-enable-baf Enable or disable B-allele
frequency output. The
default is true (enabled).
--vc-enable-baf true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
285
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-enable-decoy-
contigs
Enable/disable variant
calls on decoy contigs.
Default is false.
--vc-enable-decoy-
contigs
true/false
vc-enable-gatk-
acceleration
Enable/disable running
variant caller in
GATKmode.
--vc-enable-gatk-
acceleration
true/false
vc-enable-liquid-
tumor-mode
Enable/disable liquid
tumor mode, which
takes account of tumor-
in-normal
contamination. Default is
false (disabled).
--vc-enable-liquid-
tumor-mode
true/false
vc-enable-non-
homref-normal-filter
Enable/disable the non-
homref normal filter for
filtering out somatic
variants if the normal
sample genotype is not
homozygous reference.
Default is true (enabled).
--vc-enable-non-
homref-normal-filter
true/false
vc-enable-
orientation-bias-filter
Enable/disable the
orientation bias filter.
--vc-enable-
orientation-bias-filter
true/false
vc-enable-phasing Enable variants to be
phased when possible.
Default is true.
--vc-enable-phasing true/false
vc-enable-roh Enable or disable ROH
caller and output.
Default is true (enabled).
--vc-enable-roh true/false
vc-enable-triallelic-
filter
Enable/disable the multi-
allelic filter for somatic
mode. Default is true.
--vc-enable-triallelic-
filter
true/false
vc-enable-vcf-output Enables VCF file output
during a gVCF run.
Default is false.
--vc-enable-vcf-
output
true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
286
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-forcegt-vcf Force genotyping for
Germline small variant
calling. A .vcf or .vcf.gz
file containing a list of
small variants is
required.
--vc-forcegt-vcf A .vcf or vcf.gz file
specifying the
small variants to
force genotype.
vc-gvcf-gq-bands Define GQ bands for
gVCF output. Default is
10 20 30 40 60 80.
--vc-gvcf-gq-bands
vc-hard-filter Boolean expression for
filtering variant calls.
Default expression is:
DRAGENHardQUAL:all:
QUAL <
10.4139;LowDepth:all:
DP < 1
Parameters in the
expression can
include QD, MQ,
FS, MQRankSum,
ReadPosRankSum,
QUAL, DP, and GQ.
vc-max-reads-per-
active-region
Maximum number of
reads per active region
for downsampling.
Default is 10000.
--vc-max-reads-per-
active-region
vc-max-reads-per-
active-region-mito
Maximum number of
reads covering a given
active region for
mitochondrial small
variant calling. The
default is 40000.
--vc-max-reads-per-
active-region-mito
vc-max-reads-per-
raw-region
Maximum number of
reads per raw region for
downsampling; default is
30000.
--vc-max-reads-per-
raw-region
vc-max-reads-per-
raw-region-mito
Maximum number of
reads covering a
specified raw region for
mitochondrial small
variant calling. The
default is 40000.
--vc-max-reads-per-
raw-region-mito
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
287
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-min-base-qual Minimum base quality to
be considered for variant
calling. Default is 10.
--vc-min-base-qual
vc-min-call-qual Minimum variant call
quality for emitting a call.
Default is 3.
--vc-min-call-qual
vc-min-read-qual Minimum read quality
(MAPQ) to be considered
for small variant calling.
Default is 1 for germline,
3 for somatic T/N, and
20 for somatic T-only.
--vc-min-read-qual
vc-min-reads-per-
start-pos
Minimum number of
reads per start position
for downsampling.
Default is 10.
--vc-min-reads-per-
start-pos
vc-min-tumor-read-
qual
Minimum tumor read
quality (MAPQ) to be
considered for variant
calling.
vc-orientation-bias-
filter-artifacts
Artifact type to be
filtered. An artifact (or an
artifact and its reverse
compliment) cannot be
listed twice.
--vc-orientation-
bias-filter-artifacts
C/T, G/T,or C/T,
G/T, C/A
vc-remove-all-soft-
clips
If set to true, variant
caller does not use soft
clips of reads to
determine variants.
Default is false.
--vc-remove-all-
soft-clips
true/false
vc-roh-blacklist-bed Blacklist BED file for
ROH.
--vc-roh-blacklist-
bed
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
288
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-sq-call-threshold Set the SQ call threshold
to emit a call in the VCF.
The default is 3 for both
tumor-normal and
tumor-only.
--vc-sq-call-
threshold
vc-sq-filter-threshold Set the SQ filter
threshold to mark
emitted VCF calls as
filtered. The default is
17.5 for tumor-normal
and 6.5 for tumor-only.
--vc-sq-filter-
threshold
vc-systematic-noise BED file with site-
specific systematic noise
level to calculate AQ
score (systematic noise
score).
--vc-systematic-
noise
vc-systematic-noise-
filter-threshold
AQ threshold for
applying the systematic-
noise filter. Default 10 for
tumor-normal and 60 for
tumor-only.
--vc-systematic-
noise-filter-threshold
0–100. 10 for
tumor-normal, 60
for tumor-only
vc-target-bed Target regions BED file. --vc-target-bed
vc-target-bed-
padding
Can be used to pad all of
the target BED regions
with the specified value
(optional). If specified, is
used by the small variant
caller.
--vc-target-bed-
padding
vc-target-coverage Target coverage for
downsampling. Default
value is 500 for germline
and 50 for somatic
mode.
--vc-target-coverage
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
289
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
vc-target-coverage-
mito
Maximum number of
reads with a start
position overlapping any
given position for
mitochondrial small
variant calling. Default
value is 40000.
--vc-target-
coverage-mito
vc-tin-contam-
tolerance
Maximum tumor-in-
normal contamination
expected. Setting this to
a nonzero value enables
liquid tumor mode. The
default value is 0.15 if
liquid tumor mode is
enabled or 0 if it is
disabled.
--vc-tin-contam-
tolerance
CNV Caller Options
The following options are available for the CNV Caller.
Name Description
Command Line
Equivalent
Value
cnv-blacklist-bed Regions to blacklist for CNV
processing.
--cnv-blacklist-bed
cnv-cbs-alpha Significance level for the test to
accept change points. Default is
0.01.
--cnv-cbs-alpha
cnv-cbs-eta Type I error rate of the sequential
boundary for early stopping when
using the permutation method.
Default is 0.05.
--cnv-cbs-eta
cnv-cbs-kmax Maximum width of smaller
segment for permutation.
Default is 25.
--cnv-cbs-kmax
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
290
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
cnv-cbs-min-width Minimum number of markers for
a changed segment. Default is 2.
--cnv-cbs-min-width
cnv-cbs-nmin Minimum length of data for
maximum statistic
approximation. Default is 200.
--cnv-cbs-nmin
cnv-cbs-nperm Number of permutations used for
p-value computation. Default is
10000.
--cnv-cbs-nperm
cnv-cbs-trim Proportion of data to be trimmed
for variance calculations. Default
is 0.025.
--cnv-cbs-trim
cnv-counts-method Overlap method for counting
alignment.
--cnv-counts-method midpoint /
start /
overlap
cnv-enable-gcbias-
correction
Controls GC bias correction. --cnv-enable-gcbias-
correction
true/false
cnv-enable-gcbias-
smoothing
Controls smoothing of the GC
bias correction across adjacent
GC bins with an exponential
kernel. Default is true.
--cnv-enable-gcbias-
smoothing
true/false
cnv-enable-plots Enable/disable generation of
plots. Default is false.
--cnv-enable-plots true/false
cnv-enable-ref-calls When true, copy neutral (REF)
calls are included in the output
VCF.
--cnv-enable-ref-calls true/false
cnv-enable-self-
normalization
Enable/disable self-
normalization.
--cnv-enable-self-
normalization
true/false
cnv-enable-tracks Enable/disable generation of
track files that can be imported
into IGV for viewing. Default is
true.
--cnv-enable-tracks true/false
cnv-extreme-
percentile
Extreme median percentile value
at which to filter out samples.
Default is 2.5.
--cnv-extreme-
percentile
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
291
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
cnv-filter-bin-support-
ratio
Filters out a candidate event if
the span of supporting bins is
less than the specified ratio with
respect to the overall event
length. The default ratio is 0.2
(20% support).
--cnv-filter-bin-
support-ratio
cnv-filter-copy-ratio Minimum copy ratio threshold
value centered about 1.0 at which
a reported event is marked as
PASS in the output VCF file.
Default is 0.2
--cnv-filter-copy-ratio
cnv-filter-de-novo-
quality
Phred-scale threshold for calling
an event as de novo in the
proband.
--cnv-filter-de-novo-
quality
cnv-filter-length Minimum event length in bases at
which a reported event is marked
as PASS in the output VCF file.
Default is 10000.
--cnv-filter-length
cnv-filter-qual The QUAL value at which a
reported event is marked as
PASS in the output VCF file.
--cnv-filter-qual
cnv-input CNV input file instead of a BAM.
Either target.counts.gz or
tn.tsv.gz (for de novo).
--cnv-input
cnv-interval-width Width of the sampling interval for
CNV WGS processing.
--cnv-wgs-interval-
width
cnv-matched-normal Target counts file of the matched
normal sample.
--cnv-matched-normal
cnv-max-percent-
zero-samples
Threshold for filtering out targets
with too many zero coverage
samples. Default is 5%.
--cnv-max-percent-
zero-samples
cnv-max-percent-
zero-targets
Threshold for filtering out
samples with too many zero
coverage targets. Default is 5%.
--cnv-max-percent-
zero-targets
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
292
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
cnv-merge-distance Maximum segment gap allowed
for merging segments.
--cnv-merge-distance
cnv-merge-threshold The maximum segment mean
difference at which two adjacent
segments should be merged. The
segment mean is represented as
a linear copy ratio value.
--cnv-merge-
threshold
cnv-min-mapq Minimum MAPQ for alignment to
be counted.
--cnv-min-mapq
cnv-normals-file A single file to be used in the
panel of normals. Can be
specified multiple times, once for
each file.
--cnv-normals-file
cnv-normals-list A panel of normals file. --cnv-normals-list
cnv-num-gc-bins Number of bins for GC bias
correction. Each bin represents
the GC content percentage.
Default is 25.
--cnv-num-gc-bins 10 / 20 / 25/
50 / 100
cnv-ploidy The normal ploidy value. Used for
estimation of the copy number
value emitted in the output VCF
file. Default is 2.
--cnv-ploidy
cnv-qual-length-scale Bias weighting factor to adjust
QUAL estimates for segments
with longer lengths. Advanced
option that should not have to be
modified. Default is 0.9303 (2-
0.1).
--cnv-qual-length-
scale
cnv-qual-noise-scale Bias weighting factor to adjust
QUAL estimates based on
sample variance. Advanced
option that should not have to be
modified. Default is 1.0.
--cnv-qual-noise-scale
cnv-segmentation-
mode
Segmentation algorithm to
perform.
--cnv-segmentation-
mode
cbs / slm /
aslm
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
293
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Value
cnv-skip-contig-list A comma-separated list of contig
identifiers to skip when
generating intervals for WGS
analysis. The default contigs that
are skipped, if not specified, are
"chrM,MT,m,chrm".
--cnv-wgs-skip-
contig-list
cnv-slm-eta Baseline probability that the
mean process changes its value.
Default is 1e-5.
--cnv-slm-eta
cnv-slm-fw Minimum number of data points
for a CNV to be emitted. Default
is 0.
--cnv-slm-fw
cnv-slm-omega Scaling parameter modulating
relative weight between
experimental/biological variance.
Default is 0.3.
--cnv-slm-omega
cnv-slm-stepeta Distance normalization
parameter. The default value is
10000. Only valid for “HSLM”.
--cnv-slm-stepeta
cnv-target-bed A properly formatted BED file
that specifies the target intervals
to sample coverage over. For use
in WES analysis.
--cnv-target-bed
cnv-target-factor-
threshold
Percentile of panel-of-normals
medians used to filter out
targets. The default value is 1%
for whole genome processing
and 10% for targeted sequencing
processing.
--cnv-target-factor-
threshold
cnv-truncate-
threshold
Extreme outliers are truncated
based on this percent threshold.
Default is 0.1%.
--cnv-truncate-
threshold
cnv-use-somatic-vc-
vaf
Use somatic SNV VAFs from VC
to help determine purity and
ploidy.
-cnv-use-somatic-vc-
vaf
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
294
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Systematic Noise Creation Options
The following options are available to create systematic noise BED files from normal VCF files.
Name Description
Command Line
Equivalent
Value
vc-systematic-noise-
raw-input-list
List of VCFs to be used. One VCF
per line.
--vc-systematic-
noise-raw-input-list
vc-systematic-noise-
germline-vaf-
threshold
Minimal variant allele frequency
to remove potential germlines
from the systematic noise file
building. If not specified, all
variants are used.
--vc-systematic-
noise-germline-vaf-
threshold
0–1
vc-systematic-noise-
use-germline-tag
Whether to use DRAGEN internal
germline tagging to remove
potential germlines. Mutually
exclusive with --vc-systematic-
noise-germline-vaf-threshold .
--vc-systematic-
noise-use-germline-
tag
true/false
vc-systematic-noise-
method
Method to calculate the
systematic noise level across
samples.
--vc-systematic-
noise-method
mean / max
/ aggregate
Structural Variant Caller Options
Name Description
Command Line
Equivalent
Range
enable-sv Enable/disable structural variant
caller. Default is false.
--enable-sv true/false
sv-call-regions-bed Specifies a BED file containing
the set of regions to call.
Optionally, you can compress the
file in gzip or bzip format.
--sv-call-regions-bed
sv-denovo-scoring Enable/disable de novo quality
scoring for for structural variant
joint diploid calling. Pedigree file
must also be provided.
--sv-denovo-scoring
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
295
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Range
sv-forcegt-vcf Specify a VCF of structural
variants for forced genotyping.
The variants are scored and
included in the output VCF even
if not found in the sample data.
The variants are merged with
any additional variants
discovered directly from the
sample data.
--sv-forcegt-vcf
sv-discovery Enable SV discovery. This flag
can be set to false when using --
sv-forcegt-vcf to indicate that SV
discovery should be disabled and
only the forced genotyping input
should be used.
--sv-discovery true/false
sv-exome When set to true, configures the
variant caller for targeted
sequencing inputs, which
includes disabling high depth
filters. Default is false.
--sv-exome true/false
sv-output-contigs Set to true to have assembled
contig sequences output in a
VCF file. Default is false.
--sv-output-contigs true/false
sv-region Limit the analysis to a specified
region of the genome for
debugging purposes. Can be
specified multiple times to build a
list of regions.
--sv-region Must be in
the format
chr:startPos-
endPos.
CYP2D6_CommandLine_fDG
Name Description
Command
Line
Equivalent
Range
enable-cyp2d6 Enable CYP2D6 diplotyping. Default is false --enable-
cyp2d6
true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
296
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Repeat Expansion Detection Options
The following options can be set in the RepeatGenotyping section of the configuration file or on the
command line. For more information, see Repeat Expansion Detection with Expansion Hunter on page
152.
Parameter
Name
Description
Command Line
Equivalent
Range
enable Enable or disable repeat expansion
detection.
--repeat-
genotype-enable
true/false
specs The full path to the JSON file that contains
the repeat variant catalog (specification)
describing the loci to call.
--repeat-
genotype-specs
RNA-Seq Command Line Options
Name Description
Command Line
Equivalent
Range
enable-rna Enable processing of RNA-seq
data.
--enable-rna true/false
annotation-file Gene annotation file. Required
for quantification and gene-
fusion
--annotation-file, -a Path to GTF
(or .gtf.gz)
file
enable-rna-
quantification
Enable/disable RNA
quantification.
--enable-rna-
quantification
true/false
rna-quantification-
library-type
Specifies the type of the RNA-
seq library. Default is autodetect.
--rna-quantification-
library-type
IU, ISR, ISF,
U, SR, SF,
orA.
rna-quantification-gc-
bias
Correct for GC bias in fragment
counts.
--rna-quantification-
gc-bias
true/false
enable-rna-gene-
fusion
Enable/disable RNA gene fusion
calling.
--enable-rna-gene-
fusion
true/false
rna-gf-restrict-genes Ignore genes with biotype other
than protein coding or lncRNA for
gene fusions.
--rna-gf-restrict-
genes
true/false
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
297
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

UMI Options
Name Description
Command Line
Equivalent
Range
umi-library-type Batch option for correcting
UMIs.
--umi-library-type Not required. The
following are valid
options: random-
duplex / random-
simplex /
nonrandom-duplex
umi-enable Enable UMI-based read
processing.
--umi-enable true / false
umi-correction-
scheme
The methodology to use for
correcting sequencing errors
in UMIs.
--umi-correction-
scheme
lookup / random /
none / positional
umi-correction-table Specify the correction table
for lookup correction
scheme.
--umi-correction-
table
Path to table file
umi-emit-multiplicity Consensus read output type. --umi-emit-
multiplicity
both/duplex/simplex
umi-min-supporting-
reads
Number of input reads with
matching UMI and position
required to generate a
consensus read.
--umi-min-
supporting-reads
Integer ≥ 1. The
default is 2.
umi-metrics-
interval-file
Path to target regions file
used for UMI on target
metrics.
--umi-metrics-
interval-file
Path to valid BED file
umi-source The location to read UMIs
from.
--umi-source qname/bamtag/fastq
umi-fastq Path to a separate FASTQ
file with UMI sequences for
each read
--umi-fastq Path to valid FASTQ
file
umi-nonrandom-
whitelist
File listing valid nonrandom
UMIs, one per line
--umi-nonrandom-
whitelist
Path to file containing
valid nonrandom UMI
sequences
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
298
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Name Description
Command Line
Equivalent
Range
umi-fuzzy-window-
size
Collapse reads with
matching UMIs and
alignment positions +/ this
distance
--umi-fuzzy-
window-size
Integer ≥ 1. The
default is 3.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
299
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Technical Assistance
For technical assistance, contact Illumina Technical Support.
Website: www.illumina.com
Email: techsupport@illumina.com
Illumina Technical Support Telephone Numbers
Region Toll Free International
Australia +61 1800 775 688
Austria +43 800 006249 +43 1 9286540
Belgium +32 800 77 160 +32 3 400 29 73
Canada +1 800 809 4566
China +86 400 066 5835
Denmark +45 80 82 01 83 +45 89 87 11 56
Finland +358 800 918 363 +358 9 7479 0110
France +33 8 05 10 21 93 +33 1 70 77 04 46
Germany +49 800 101 4940 +49 89 3803 5677
Hong Kong, China +852 800 960 230
India +91 8006500375
Indonesia 0078036510048
Ireland +353 1800 936608 +3531 695 0506
Italy +39 800 985513 +39 236003759
Japan +81 0800 111 5011
Malaysia +60 1800 80 6789
Netherlands +31 800 022 2493 +31 20 713 2960
New Zealand +64 800 451 650
Norway +47 800 16 836 +47 21 93 96 93
Philippines +63 180016510798
Singapore 1 800 5792 745
South Korea +82 80 234 5300
Spain +34 800 300 143 +34 911 899 417
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
300
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide

Region Toll Free International
Sweden +46 2 00883979 +46 8 50619671
Switzerland +41 800 200 442 +41 56 580 00 00
Taiwan, China +886 8 06651752
Thailand +66 1800 011 304
United Kingdom +44 800 012 6019 +44 20 7305 7197
United States +1 800 809 4566 +1 858 202 4566
Vietnam +84 1206 5263
Safety data sheets (SDSs)—Available on the Illumina website at support.illumina.com/sds.html.
Product documentation—Available for download from support.illumina.com.
Document # 1000000141465 v00
For Research Use Only. Not for use in diagnostic procedures.
301
Illumina DRAGEN Bio-IT Plaform 3.7 User Guide
