
GUIDANCE FOR SPONSORS
Lot Release Program for Schedule D (Biologic) Drugs
Published by authority of the
Minister of Health
Date Adopted 2005/03/23
Effective Date 2005/06/01
Health Products and Food Branch

Our mission is to help the people of Canada
maintain and improve their health.
Health Canada
HPFB’s Mandate is to take an integrated approach to the
management of the risks and benefits to health related to
health products and food by:
• Minimizing health risk factors to Canadians while
maximizing the safety provided by the regulatory
system for health products and food; and,
• Promoting conditions that enable Canadians to
make healthy choices and providing information so
that they can make informed decisions about their
health.
Health Products and Food Branch
THERAPEUTIC PRODUCTS DIRECTORATE / BIOLOGICS AND GENETIC THERAPIES
DIRECTORATE WEBSITE (S)
LET YOUR COMPUTER DO THE SEARCHING!
... Need to know how to market a new drug in Canada?
... Want information on the drug regulatory process?
... Need to know what the newest drugs on the
Canadian market are?
... Want direct access to forms and policies?
... Need to know the requirements for labelling drugs?
All this and more is available on the
Therapeutic Products Directorate / Biologics and Genetic Therapies Directorate Website (s)
at
www.hc-sc.gc.ca/hpfb-dgpsa/tpd-dpt/
www.hc-sc.gc.ca/hpfb-dgpsa/bgtd-dpbtg
© Minister of Public Works and Government Services Canada 2005
Available in Canada through
Health Canada - Publications
Brooke Claxton Building, A.L. #0913A
Tunney's Pasture
Ottawa, Ontario
K1A 0K9
Tel: (613) 954-5995
Fax: (613) 941-5366
Également disponible en français sous le titre: Ligne directrice à l’intention des promoteurs: Programme
d’Autorisation de Mise en Circulation des Lots de Drogues Visées à l’Annexe D (Produits Biologiques)
Catalogue No. E
ISBN

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
FOREWORD
Guidance documents are meant to provide assistance to industry and health care professionals on
how to comply with the policies and governing statutes and regulations. They also serve to provide
review and compliance guidance to staff, thereby ensuring that mandates are implemented in a fair,
consistent and effective manner.
Guidance documents are administrative instruments not having force of law and, as such, allow for
flexibility in approach. Alternate approaches to the principles and practices described in this
document may be acceptable provided they are supported by adequate scientific justification.
Alternate approaches should be discussed in advance with the relevant program area to avoid the
possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to
request information or material, or define conditions not specifically described in this guidance, in
order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic
product. Health Canada is committed to ensuring that such requests are justifiable and that
decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant
sections of other applicable guidances.

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
TABLE OF CONTENTS
1.0 INTRODUCTION ..................................................................2
1.1 Purpose .....................................................................2
1.2 The Lot Release Program ......................................................2
1.3 Scope ......................................................................3
1.4 Abbreviations, Acronyms, and Definitions .........................................3
2.0 EVALUATION GROUPS ............................................................4
2.1 Group 1: Pre-Approval Stage ................................................... 4
2.2 Group 2 to 4: Post-Approval Stage ...............................................5
3.0 FACTORS CONSIDERED DURING ASSIGNMENT OF PRODUCTS TO EVALUATION GROUPS 6
3.1 Product Indication ............................................................6
3.2 Nature of the Product ..........................................................6
3.3 Production History ............................................................6
3.4 Inspection History ............................................................7
3.5 Testing History .............................................................. 7
3.6 Post-market Experience ........................................................7
4.0 MOVEMENT BETWEEN EVALUATION GROUPS ...................................... 7
5.0 SPONSOR INFORMATION AND REGULATORY REQUIREMENTS .......................8
5.1 Yearly Biologic Product Report .................................................. 9
6.0 HUMAN-DERIVED EXCIPIENTS ...................................................10
7.0 APPEALS ........................................................................10
8.0 EFFECTIVE DATE ................................................................ 11
9.0 ADDITIONAL INFORMATION ......................................................11
APPENDIX I: Fax-Back Forms ..........................................................12
APPENDIX 1A: Clinical Trial Material(s) ..............................................12
APPENDIX 1B: Group 4 Products Containing Human-derived Excipients .....................13
APPENDIX 1C: Group 4 Products ....................................................14
APPENDIX II: Targeted Testing ..........................................................15
APPENDIX III: Periodic Testing ..........................................................16
APPENDIX IV: Summary of Requirements for Evaluation Groups ............................... 17
Effective Date: 2005/06/01 1

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
1.0 INTRODUCTION
1.1 Purpose
The purpose of this document is to outline the Lot Release Program for Schedule D (biologic) drugs
and the extent of the review and testing of biologic drugs prior to their release for sale in Canada by
the Biologics and Genetic Therapies Directorate (BGTD).
1.2 The Lot Release Program
Each lot of a Schedule D (biologic) drug is subject to the Lot Release Program before sale
1
in
Canada. The risk-based Lot Release Program covers both pre- and post-market stages. The Lot
Release Program derives its legislative authority from section C.04.015 of the Food and Drug
Regulations
2
. Products are assigned to one of four Evaluation Groups, with each group having
different levels of regulatory oversight (testing and/or protocol review) based on the degree of risk
associated with the product. The graduated risk-based approach to testing and oversight allows
BGTD to focus ongoing testing on products for which enhanced surveillance is indicated such as
vaccines and blood products. The criteria used to determine the appropriate Evaluation Group
include, but are not limited to, the nature of the product, the target population, the lot testing history
in BGTD, and the manufacturer’s production and testing history.
In general, the outcome of testing and/or protocol review is communicated to the manufacturer via a
Release Letter prior to the product’s release for sale in Canada. In certain situations, a Fax-back
process is used. A Fax-back form (Appendix I) which is submitted by the manufacturer
3
attests that
all specifications have been met; receipt is acknowledged by Fax-back within 48 hours.
1
“Sell” includes offer for sale, expose for sale, have in possession for sale and distribute, whether or not
the distribution is made for consideration.
2
C.04.015 On written request from the Director, every fabricator, packager/ labeller, tester, distributor
referred to in paragraph C.01A.003(b) and importer of a drug shall submit protocols of tests together
with samples of any lot of the drug before it is sold, and no person shall sell any lot of that drug if the
protocol or sample fails to meet the requirements of these Regulations
C.01A.003 b) a distributor of a drug for which that distributor holds the drug identification number.
3
“Manufacturer” refers to the person in Canada responsible for the sale of the drug which may include, but is
not limited to, the establishment licence holder, fabricator, DIN holder, and/or distributor of a drug for which
that distributor holds the DIN.
Effective Date: 2005/06/01 2

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
1.3 Scope
1.3.1 This guidance document is applicable to all Schedule D (biologic) drugs regulated by
BGTD.
1.3.2 In this guidance document, “shall” is used to express a requirement, i.e., a provision that
the user is obliged to satisfy in order to comply with the regulatory requirements; “should” is
used to express a recommendation or that which is advised but not required; and “may” is
used to express an option or that which is permissible within the limits of the guidance
document.
1.4 Abbreviations, Acronyms, and Definitions
1.4.1 Abbreviations and Acronyms
BGTD Biologics and Genetic Therapies Directorate
CBE Centre for Biologics Evaluation
CERB Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics
CoA Certificate of Analysis
CPID Certified Product Information Document
CTA Clinical Trial Application
DIN Drug Identification Number
GMP Good Manufacturing Practices
HDE Human-derived Excipient
MRA Mutual Recognition Agreement
NDS New Drug Submission
NOC Notice of Compliance
RAD Regulatory Affairs Division
S/NDS Supplemental New Drug Submission
YBPR Yearly Biologic Product Report
1.4.2 Definitions
Aborted Lot
Wherein a batch record is issued, for any step in the production of a bulk intermediate or
final product, but the batch is not released due to a breakdown in the manufacturing
process related to facility systems, equipment, or procedures.
Consistency Testing
Laboratory testing performed by BGTD during the review period for New Drug
Submissions or Supplemental New Drug Submissions. Generally, samples from 3 to 5
consecutively manufactured lots are tested.
Effective Date: 2005/06/01 3

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
Human-derived Excipient
Any component of a drug product derived from a human source, other than
the claimed therapeutic ingredient(s).
Lot Failure
A drug substance or final product lot or batch that has been rejected due to failure to
meet in-process or final release specifications.
Protocol of Tests
Information submitted by the manufacturer required to demonstrate that the lot is
acceptable for sale in Canada. This may include Certificates of Analysis, attestations,
and completed worksheets.
Reprocessing
Subjecting all or part of a batch or lot of an in-process drug, a bulk process intermediate
(final biological bulk intermediate) or a bulk drug of a single batch/lot to a previous step
in the validated manufacturing process due to failure to meet predetermined
specifications. Reprocessing procedures are foreseen as occasionally necessary and are
validated and pre-approved as part of the marketing authorization.
Reworking
Subjecting an in-process drug, a bulk process intermediate (final biological bulk
intermediate), or final product of a single batch/lot to an alternate manufacturing process
due to a failure to meet predetermined specifications. Reworking is an unexpected
occurrence and is not pre-approved as part of the marketing authorization.
Yearly Biologic Product Report
A report required every year for all approved Schedule D (biologic) drugs.
2.0 EVALUATION GROUPS
2.1 Group 1: Pre-Approval Stage
All products under review as a Clinical Trial Application (CTA), or New Drug Submission (NDS),
and in some cases a Supplementary New Drug Submission (S/NDS), are assigned to Evaluation
Group 1 during the review period. Group 1 has two distinct sub-groups.
2.1.1 Group 1A: Clinical Trial Materials
This Evaluation Group consists of clinical trial materials associated with authorized CTAs.
Sponsors are required to complete and file a Fax-back form (Appendix IA) and await a signed
response from BGTD prior to use of the clinical trial material. For prophylactic vaccines,
Effective Date: 2005/06/01 4

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
BGTD issues a formal release letter for use of the vaccine lot in the clinical trial; the protocol
of tests and usually samples are required to be submitted to BGTD.
2.1.2 Group 1B: Consistency Testing
This Evaluation Group is intended for consistency samples associated with an NDS or S/NDS.
Generally, samples from 3 to 5 consecutively manufactured lots are tested by BGTD to ensure
consistency of the manufacturing process. Upon request, consistency lots may be released for
sale in Canada once an NOC is issued; a formal release letter is required from BGTD.
2.2 Group 2 to 4: Post-Approval Stage
Evaluation Groups 2 to 4 apply to biologic products for which an NOC has been issued.
2.2.1 Group 2: Sample Testing and Protocol Review
Products requiring the highest level of assessment after issuance of an NOC are assigned to
this Evaluation Group. Products in this group are subjected to Targeted Testing (Appendix II).
A formal Release Letter which approves the sale of the lot in Canada is required from BGTD
before each lot is sold. The targeted timeframe for products in this Group to be released is 6
weeks after receipt of all required information and samples. The timeframe for some products,
such as those with long bioassays, may be longer. Expedited release may be granted in
exceptional cases and upon appropriate justification (such as product shortage in Canada).
2.2.2 Group 3: Protocol Review and Periodic Testing
Products requiring a moderate level of assessment after issuance of an NOC are assigned to
this Evaluation Group. A formal Release Letter which approves the sale of the lot in Canada
is required from BGTD before each lot is sold. For products in this Group, BGTD reviews
testing protocols but samples are not routinely submitted by the manufacturer for Targeted
Testing. Instead, at the discretion of BGTD, samples may be requested for Periodic Testing
(Appendix III). The targeted timeframe for products in this Group to be released for sale is
two weeks from the date that all required information is received.
2.2.3 Group 4: Notification and Periodic Testing
Products in this Evaluation Group do not undergo sample testing or protocol review by
BGTD. When a Schedule D (biologic) drug has been assigned to Evaluation Group 4, the
manufacturer of that drug is required to notify BGTD via Fax-back (Appendix I) when a lot is
to be sold in Canada. A Release Letter is not required prior to sale. At the discretion of
BGTD, products in Evaluation Group 4 may also be subjected to Periodic Testing (Appendix
III).
Effective Date: 2005/06/01 5

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
3.0 FACTORS CONSIDERED DURING ASSIGNMENT OF PRODUCTS TO
EVALUATION GROUPS
The factors considered when assigning products to Evaluation Groups are outlined in sections 3.1 to
3.6.
3.1 Product Indication
The degree of oversight to which a Schedule D (biologic) drug is subjected to during Lot Release is
associated with its indication and risk/benefit assessment. Considerations include the following:
• age of target population (e.g. infants, seniors etc.)
• disease state being treated (e.g. life threatening, acute, chronic)
• duration of treatment (e.g. short/long term)
• health status (e.g. incurable, healthy)
• objective (e.g. treatment vs prevention vs replacement vs diagnostic)
• population size (limited/widespread use)
3.2 Nature of the Product
All Schedule D (biologic) drugs are assessed as to their nature, which is a consideration for
Evaluation Group assignment. Considerations in evaluating the nature of the product include the
following:
• source and level of control of the raw materials
• complexity, robustness and level of control of the manufacturing process
• chemical complexity of the drug substance
• chemical complexity of the drug product
• reliability and complexity of the methods used to evaluate identity, purity, and potency of
the drug substance and the drug product
3.3 Production History
Consistency of manufacturing and the ability to consistently produce a drug without reworking is a
consideration in the assignment of products to Evaluation Groups.
3.3.1 Lot Failures and Aborted Lots
Information on the incidence of lot failures and severity of cause of aborting a lot during
production contributes to the assignment of a product to an Evaluation Group.
3.3.2 Reprocessed Lots
Changes in the incidence and extent of reprocessing are an indication of the degree of control
in the manufacturing process and contributes to the information used for the assignment of
products to Evaluation Groups.
Effective Date: 2005/06/01 6

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
3.4 Inspection History
Quality and safety issues found during On-Site Evaluations and other inspections contribute to the
assignment of products to the Evaluation Groups.
3.5 Testing History
The test results submitted by or for the manufacturer, as well as test results obtained by BGTD are
also part of the considerations used in assigning products to an Evaluation Group. Additional data
may be derived from test protocol review during inspection and the exchange of inspection reports
through Mutual Recognition Agreements (MRA), and other sources. In addition to actual test
results, the rate of re-test due to test failures and invalid tests is also considered.
3.6 Post-market Experience
Information from adverse drug reaction reports, product complaints, product recalls, and
withdrawals contribute to the post-market safety profile of the drug product. This information is
also used in the assignment of products to Evaluation Groups.
4.0 MOVEMENT BETWEEN EVALUATION GROUPS
The initial assignment of a product to an Evaluation Group upon receiving approval is at the
discretion of BGTD, taking into account considerations outlined in Section 3. Usually, with the
exception of vaccines which may remain in Evaluation Group 2 indefinitely, products assigned to
Evaluation Group 2 remain in that group for a period of one year, or until such time as five lots have
been tested and released, whichever is longest. Following the one year or period where five lots
have been tested satisfactorily, the product may be re-assigned into Evaluation Group 3 or 4
providing there have been consistent and reliable testing outcomes observed while in Group 2 and
that there have been no changes in the manufacturing process that may have an impact on the
quality of the drug.
Products that are produced from well controlled raw materials through reliable and consistent
processes, and that can be readily assessed with respect to identity, purity and potency through
reliable test protocols may be assigned to Evaluation Group 4 at the time of approval.
Movement through the Evaluation Groups may be bi-directional. For example, quality issues
detected by Periodic Testing during Evaluation Groups 3 and 4 may result in product re-assignment
to Group 2 or 3 until such time that sufficient evidence to support re-assignment of the product is
available. Information obtained during routine inspections or from other sources may also affect the
Evaluation Group assignment.
Re-assignment of a product to a different Evaluation Group occurs in one of two ways:
1) Upon review of the Yearly Biologic Product Report, manufacturers are notified if the product
Effective Date: 2005/06/01 7

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
is re-assigned to a different Group. The manufacturer may appeal within 60 days of
notification, extensions may be given on a case-by-case basis upon request by the
manufacturer, see Section 7.0.
2) Manufacturers may apply for re-assignment, in writing and provide the data outlined in
Section 5.1 to the Director of CBE or CERB in order to be assessed for re-assignment into a
different Evaluation Group.
5.0 SPONSOR INFORMATION AND REGULATORY REQUIREMENTS
Under section C.04.015
1
of the Food and Drug Regulations, the manufacturer shall provide
information supporting Lot Release. A summary of the information requirements for the different
Evaluation Groups is provided in Appendix IV.
Manufacturers of Schedule D (biologic) drugs in Evaluation Groups 2, 3, and 4 shall provide, under
section C.01.014.5
4
, C08.007
5
and/or C.08.008 of the Food and Drug Regulations, information
annually to Health Canada (BGTD). For the Lot Release Program, a Yearly Biologic Product
Report (YBPR) is required (See section 5.1). Information from the YBPR may be used to verify the
consistency of the process, to assess the on-going safety and quality of the product, and to highlight
any trends. The information may also be part of the consideration of re-assignment of a product into
a different Evaluation Group.
4
Every manufacturer of a drug shall, annually before the first day of October and in a form authorized by the
Director, furnish the Director with a notification signed by the manufacturer or by a person authorized to
sign on his behalf, confirming that all the information previously supplied by the manufacturer with respect
to that drug is correct.
5
Where a manufacturer has received a notice of compliance issued in respect of a New Drug Submission or
Abbreviated New Drug Submission or a supplement to either submission, the manufacturer shall establish
and maintain records, in a manner that enables an audit to be made, respecting
a) animal or clinical experience, studies, investigations and tests conducted by the manufacturer or
reported to him by any person concerning that new drug;
b) reports from the scientific literature or the bibliography there from that are available to him
concerning that new drug;
c) experience, investigations, studies and tests involving the chemical or physical properties or any other
properties of that new drug;
d) any substitution of another substance for that new drug or any mixing of another substance with that
new drug;
e) any error in the labelling of that new drug or in the use of the labels designed for that new drug;
f) any bacteriological or any significant chemical or physical or other change or deterioration in any lot
of that new drug;
g) any failure of one or more distributed lots of the new drug to meet the specifications established for
that new drug in the submission or supplement; and
(h) any unusual failure in efficacy of that new drug.
Effective Date: 2005/06/01 8

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
5.1 Yearly Biologic Product Report
5.1.1 The following information should be part of the YBPR:
5.1.1.1 Production information on both drug substance and drug product lots:
• number of lots produced for or sold on the Canadian market
• number of lots produced or sold internationally, from facilities licensed to produce
lots for Canada
• number of lots reprocessed from facilities licensed to produce lots for Canada
• a review of all commercial lots intended for Canadian and international use that failed
to meet established specifications, or were aborted due to manufacturing process
failures, including those having been determined as having failed through studies,
investigations and tests conducted by the manufacturer or reported to him by any
person
• a review of all changes carried out to the process or analytic methods
• a concise, high-level review of critical deviations or non-conformances, related
investigations, and resolution/corrective actions
• a list of changes to raw material suppliers and changes to non-compendial
specifications
5.1.1.2 Information on drug substance and drug product test methods:
• frequency of retesting due to out-of-specification, including clarification on the
reason for retesting
• frequency of invalid tests for stability-indicating test methods
5.1.1.3 Information on drug substance and drug product test results:
• a review of critical in-process controls and finished product results
• trend analysis for stability-indicating test methods
• a review of results of ongoing stability program(s)
5.1.1.4 Facility information:
• a review of regulatory actions taken by competent authorities that affect GMP status
5.1.1.5 Analysis of Adverse Drug Reaction Reports (Canadian and International) received
by the manufacturer attributable to product quality
5.1.1.6 All product recalls including the reason for the recall and a summary of any
corrective actions taken
5.1.1.7 If changes affecting the CPID have been made, an updated CPID (annotated and
non-annotated, hard copy and electronic) is to be provided with the YBPR
Effective Date: 2005/06/01 9

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
5.1.2 Submission of Yearly Biologic Product Report
A report prepared for another competent regulatory authority that contains the information
outlined in sections 5.1.1.1 to 5.1.1.7 may be updated with Canadian-specific information and
submitted as the YBPR.
The YBPR should be submitted as an Addendum to the Annual Drug Notification Report no
later than October of each year.
Alternatively, the date of first submission may be negotiated with BGTD, after which
subsequent reports will be submitted every 12 months to the Regulatory Affairs Division
(RAD).
6.0 HUMAN-DERIVED EXCIPIENTS
For Schedule D (biologic) drugs containing human-derived excipients (HDE), the sponsor shall
maintain a traceable link between the drug product and the lot number(s) of the HDE used for the
drug product lot
6
.
The lot number, manufacturer, and other pertinent HDE information must be provided as part of the
documentation for the lot for release of products in Evaluation Groups 2 and 3, or via a Fax-back
form at time of sale in Canada for products in Evaluation Group 4.
Products in Evaluation Group 1A formulated with HDE must be supported by CTA Fax-back
forms.
All HDE used as excipients must meet the regulatory requirements for approval of HDEs as
specified by BGTD. Any changes to the manufacturing of HDEs should be appropriately filed to
BGTD.
7.0 APPEALS
Upon the assignment of a product into an Evaluation Group, manufacturers may appeal the
grouping of their product in writing to the Director of CBE or CERB. BGTD will target 60
calendar days to assess the submitted information and provide a written response to the
Interpretation 11 of regulations C.02.011 and C.02.012 as outlined in the GMP Annex for Schedule D
drugs, Part 1, indicates that "Batch records must document all biological starting materials and in-
process materials used, in addition to all relevant test results."
Effective Date: 2005/06/01
6
10

Health Canada Lot Release Program for Schedule D (Biologic) Drugs
Guidance for Sponsors
manufacturer. Similarly, requests for re-assignment to another Evaluation Group should be directed
to the Director of CBE or CERB.
8.0 EFFECTIVE DATE
This Guidance document is effective as of June 1, 2005.
The requirements for sponsors to submit YBPRs and to notify BGTD via Fax-back for all Group 4
products become effective June 1, 2006. Until then, the Fax-back process for Group 4 products will
continue to apply only to those products that contain HDE.
9.0 ADDITIONAL INFORMATION
If you have any questions or require information regarding this guidance, contact:
Regulatory Affairs Division
Centre for Policy and Regulatory Affairs
Biologics and Genetic Therapies Directorate
Telephone: (613) 957-1722
Fax: (613) 941-1708
E-mail: [email protected]a
Effective Date: 2005/06/01 11
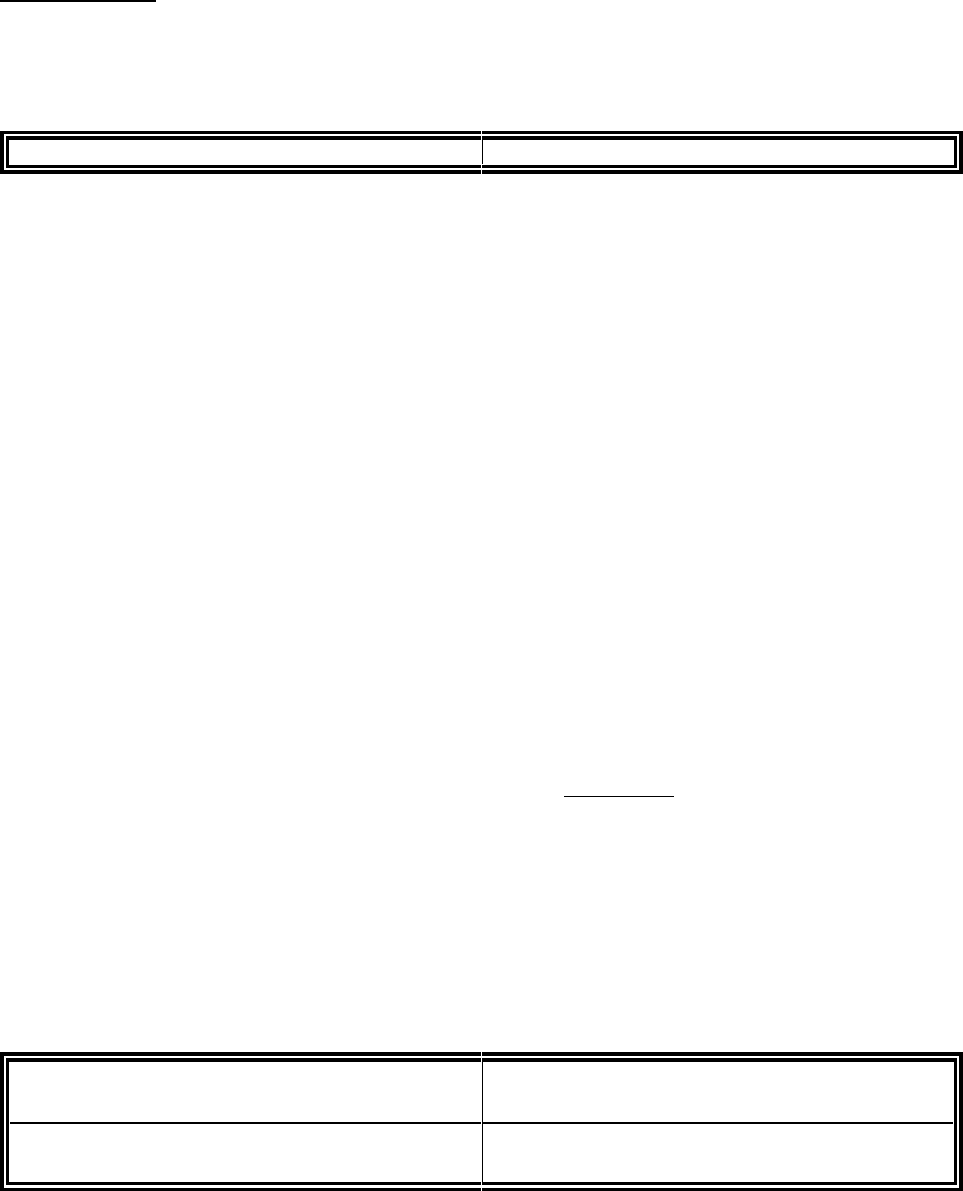
APPENDIX IA
CTA FAX-BACK FORM
CLINICAL TRIAL MATERIAL(S)
FAX COMPLETED FORM TO REGULATORY AFFAIRS DIVISION
Fax: (613) 941-1708
Date Received: Tracking #:
SUBMISSION INFORMATION (Manufacturer to complete this Section, for each drug lot)
Clinical Trial Application (CTA) Control #:
Proper Name:
Trade Name:
Manufacturer:
Sponsor:
Protocol(s) #:
Date of CTA Clearance:
DRUG PRODUCT INFORMATION
Lot Number:
Strength/Presentation:
Date of Manufacture:
Current Expiry/Retest:
Drug Substance Batch Number(s):
Current Expiry/Retest Date:
Human-derived Excipient Product Name/Concentration:
Human-derived Excipient Manufacturer:
Human-derived Excipient Plasma Source: a) Country of Origin b) Recovered � Apheresis �
Human-derived Excipient Lot Number(s):
This certifies that, all release tests for the above Drug Substance and Drug Product lot(s) have been
completed as outlined in the above submission; the source and testing of any associated human-derived
excipients are consistent with the approved submission; and (check one box only),
� All test results are within approved specifications; or,
� Not all testing specifications have been met - testing protocol, explanation, and rationale for
use are appended.
Responsible Head, or Designate:_____________________________ Phone: ______________________
Fax to: Fax No:
Signature: Date Fax-Back Complete:
If you do not receive all pages or this transmission is not clear please contact (613) 957-1722.
Rev. 1 Rev. Date: March 18, 2005
Effective Date: June 1, 2005
12

APPENDIX IB
FAX-BACK FORM
GROUP 4 PRODUCTS CONTAINING HUMAN-DERIVED EXCIPIENTS
FAX COMPLETED FORM TO REGULATORY AFFAIRS DIVISION
Fax: (613) 941-1708
Date Received: Tracking #:
DRUG PRODUCT (Manufacturer to complete this Section, for each drug lot)
Product Name/Concentration:
Trade Name (if applicable):
Drug Identification Number (DIN):
Manufacturer:
Lot Number:
Date of Manufacture/Expiry Date:
ASSOCIATED HUMAN-DERIVED EXCIPIENT(S)
Product Name/Concentration:
Trade Name (if applicable):
Manufacturer:
DIN (if applicable):
Plasma Source: a) Country of Origin b) Recovered � Apheresis �
Lot Number:
Date of Manufacture/Date of Expiry:
This certifies that the source and testing of the human-derived excipient identified above, and associated
with the drug product lot identified above, is consistent with the approved submission for the drug product
and/or with subsequent agreements made with the Centre for Biologics Evaluation (CBE) or the Centre for
Evaluation of Radiopharmaceuticals and Biotherapeutics (CERB)
Responsible Head, or Designate:__________________________________ Phone: _________________
Fax to: Fax No:
Signature: Date Fax-Back Complete:
If you do not receive all pages or this transmission is not clear please contact (613) 957-1722.
Rev. 1 Rev. Date: March 18, 2005
Effective Date: June 1, 2005
13

APPENDIX IC
FAX-BACK FORM
GROUP 4 PRODUCTS
FAX COMPLETED FORM TO REGULATORY AFFAIRS DIVISION
Fax: (613) 941-1708
Date Received: Tracking #:
DRUG PRODUCT (Manufacturer to complete this Section, for each drug lot)
Product Name/Concentration:
Trade Name (if applicable):
Drug Identification Number (DIN):
Manufacturer:
Lot Number:
Date of Manufacture/Expiry Date:
ASSOCIATED HUMAN-DERIVED EXCIPIENT(S) Check one: � YES � NO
Product Name/Concentration:
Trade Name (if applicable):
Manufacturer:
DIN (if applicable):
Plasma Source: a) Country of Origin b) Recovered � Apheresis �
Lot Number:
Date of Manufacture / Date of Expiry:
This certifies that the source and testing of the human-derived excipient identified above, and associated
with the drug product lot identified above, is consistent with the approved submission for the drug product
and/or with subsequent agreements made with the Centre for Biologics Evaluation (CBE) or the Centre for
Evaluation of Radiopharmaceuticals and Biotherapeutics (CERB)
Responsible Head, or Designate:______________________________________ Phone: ______________
Fax to: Fax No:
Signature: Date Fax-Back Complete:
If you do not receive all pages or this transmission is not clear please contact (613) 957-1722.
Rev. 1 Rev. Date: March 18, 2005
Effective Date: June 1, 2006
14

APPENDIX II: TARGETED TESTING
Targeted testing specifies a combination of one or more assays, applied to all lots, for a particular
biologic product. It may include a subset of tests ranging from one (1) to the complete set of assays
proposed by the manufacturer in the submission of a CTA, NDS, or S/NDS. Routine lot release
testing may be restricted to those critical assays in which failure to meet approved specifications
may reflect product quality or safety. Certificates of Analyses are also reviewed. The targeted
timeframe for products in this Group to be released is 6 weeks after receipt of all required
information and samples. Certain products with lengthy bioassays may take longer.
The targeted testing regime developed for Group 2 products is based on:
a) Risk assessment of each test as determined by i) the possibility of an incorrect test result,
and ii) the risk to safety associated with an incorrect test result
b) BGTD's experience with testing of the product and results of testing of consistency lots
and/or clinical trial materials
c) Lot failure information provided by the manufacturer
d) Other pertinent information such as information obtained from other Regulatory Agencies,
Product Recalls, and Adverse Drug Reaction reports
For products that require lengthy release tests (e.g. animal testing), concurrent testing by BGTD and
the manufacturer will be considered. In the case of concurrent testing, prompt notification of any
testing failures must be provided to BGTD.
15

APPENDIX III: PERIODIC TESTING
Products in Evaluation Groups 3 and 4 are subject to Periodic Testing. Lot samples may be
requested by BGTD for Periodic Testing to confirm that the product meets specifications. Lot
samples are selected based on production history, inspection history, testing history and other
related factors.
If lot samples have been requested by BGTD for Periodic Testing, the targeted timeframe for release
is 6 weeks from the date that all required information is received.
16
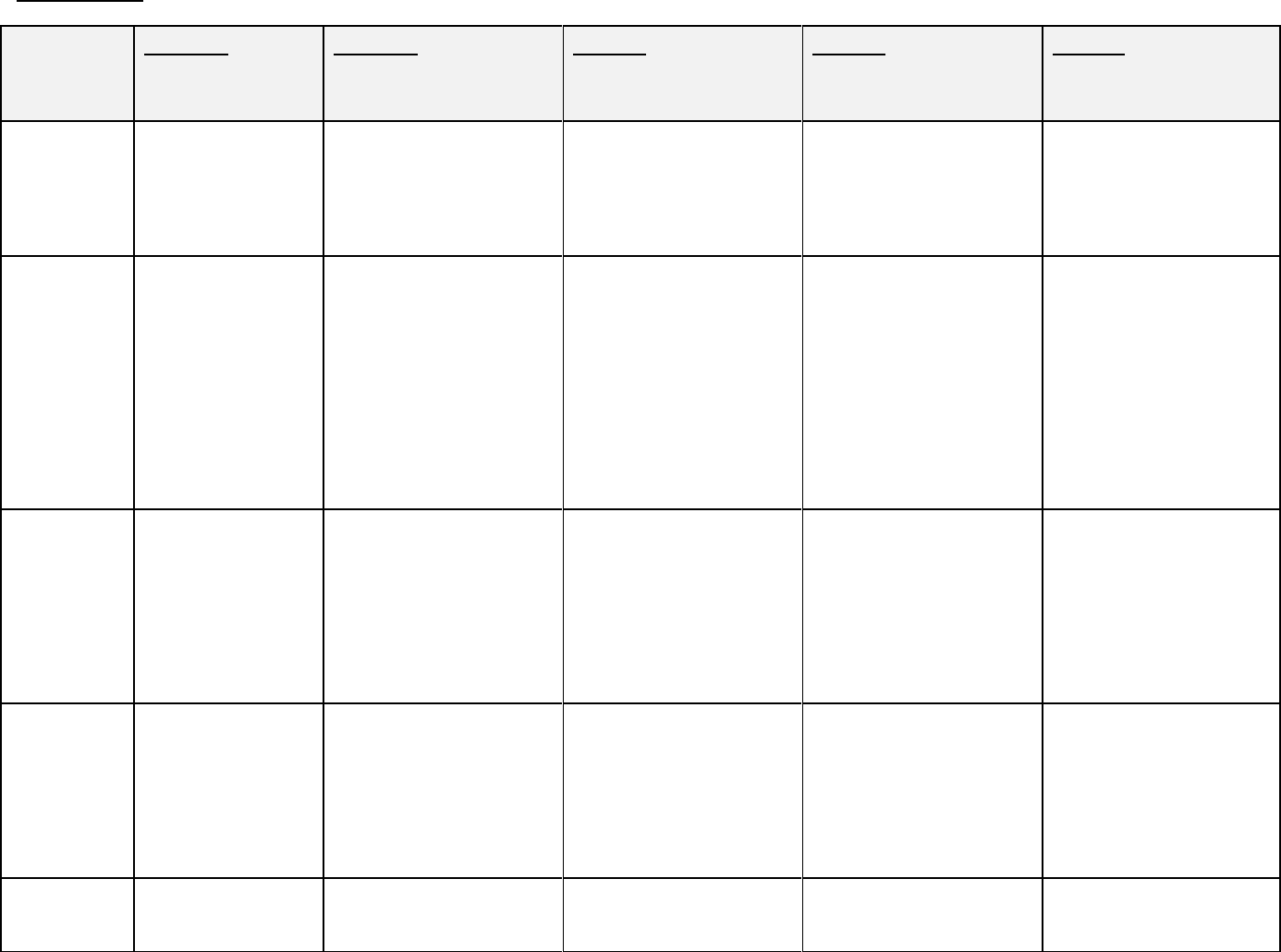
APPENDIX IV:
SUMMARY OF REQUIREMENTS FOR EVALUATION GROUPS
Evaluation GROUP 1A GROUP 1B GROUP 2 GROUP 3 GROUP 4
Group Pre-Approval Pre-Approval Post-Approval Post-Approval Post-Approval
Description Clinical Trials Associated with NDS or S/NDS
submissions.
Products requiring the highest
level of assessment.
Products requiring a moderate
level of assessment.
Products requiring a low level
of assessment.
Sample Prophylactic Vaccines: Samples from 3 to 5 Targeted Testing (mandatory Products in Group 3 are subject Products in Group 4 are subject
Requirements Samples submitted for
testing by BGTD
Other Biologics:
No samples required
consecutively manufactured lots
are submitted to BGTD for lot-
to-lot consistency testing
submission of samples of all
lots to BGTD for testing). See
Appendix II.
to Periodic Testing. See
Appendix III.
to Periodic Testing. See
Appendix III.
The manufacturer must notify
BGTD on an annual basis of
lots sold in Canada
Document Prophylactic Vaccines: Submission of Protocols of tests Submission of Protocols of tests Submission of Protocols of tests Fax-back form with lot number
Requirements Submission of Protocols
of tests and/or CoAs to
BGTD for review
Other Biologics:
Sponsors are required to
complete and file a Fax-
back form, which must
include a rationale if
testing specifications
have not been met
and/or CoAs to BGTD for
review
and/or CoAs to BGTD for
review
and/or CoAs to BGTD for
review
of product at time of sale in
Canada plus information on
HDE’s if product contains HDE
Approval Prophylactic Vaccines: Upon request, lots from which A written approval for sale in A written approval for sale in Not Applicable
Mechanism A written approval in
the form of a release
letter is required
Other Biologics:
Fax-back form will be
returned by fax to the
sponsor
consistency samples were taken
may be released for sale in
Canada once an NOC is issued
A written approval in the form
of a release letter is required for
all lots
the form of a release letter is
required for all lots
the form of a release letter is
required for all lots
Target Not Applicable Not Applicable 6 weeks after receipt of all 2 weeks after receipt of all If Periodic Testing samples are
Timeline required information and
samples
Expedited release may be
granted in exceptional cases and
upon appropriate justification
such as product shortage
required information
If Periodic Testing samples are
requested by BGTD, the target
timeline is 6 weeks
requested by BGTD, the target
timeline is 6 weeks
Reporting
Requirements
Not Applicable Not Applicable Yearly Biologic Product Report Yearly Biologic Product Report Yearly Biologic Product Report
17

BGTD Lot Release Working Group Members
Guidance for Sponsors: Lot Release Program
For Schedule D (Biologic) Drugs
Name Centre
Jacqueline Fildes Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics
Sylvia Frenette Centre for Biologics Evaluation
Basanti Ghosh Centre for Biologics Evaluation
Nancy Green Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics
Stephanie Hardy Centre for Policy and Regulatory Affairs
Brenda Moffitt Centre for Biologics Evaluation
Kwasi Nyarko Centre for Policy and Regulatory Affairs
Harold Rode Centre for Biologics Evaluation
Lori Schauland Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics
Willem Stevens Centre for Biologics Evaluation
Vincent Tong Centre for Policy and Regulatory Affairs
Julie Wallace Centre for Policy and Regulatory Affairs
KwokHim Yeung Centre for Biologics Evaluation
Members of the Working Group who contributed to the original Draft Lot Release Guidance:
Tara Bower, Jacquie Fildes, Sylvia Frenette, Brenda Moffitt, Sharon Mullin, Jean Peart, Mary Podnar
(lead), and Walter Yarosh.
18
